Introduction, présentation du sujet et des objectifs de cette étude
Depuis le dramatique accident lié à l’utilisation de la thalidomide, les différences de réactivités biologiques des isomères optiques ont été mises en lumière. La nécessité de mener des études pharmacologiques sur les isomères séparés est apparue. Les nouvelles directives de la pharmacopée européenne de mise sur le marché, ont confirmé récemment cet impératif.
Il est donc indispensable d’obtenir chaque isomère sous une forme énantiomériquement pure. A cette fin, la synthèse asymétrique a connu un développement important. Il existe souvent des limites à l’obtention de produits optiquement purs par synthèse. Il est important de pouvoir contrôler la pureté optique des isomères obtenus par synthèse asymétrique. Il est aussi nécessaire, en particulier lorsqu’un contrôle stéréosélectif total n’a pu être réalisé, de disposer de méthodes physico-chimiques de séparation pour purifier les énantiomères.
Une
méthode utilisée pour obtenir la séparation d’énantiomère
est la réalisation de complexes diastéréoisomères
de propriétés physiques différentes rendant possible leur
séparation physique. De nombreux auteurs, à la suite de Dalgliesh
ont posé les bases théoriques de la reconnaissance chirale par
complexation. Le modèle des trois points d’interaction a été
très largement étudié, informant ainsi sur le facteur géométrique
de la reconnaissance chirale. Pirkle a, quant à lui, étudié
la question du point de vue physique,
par la mise au point de colonnes chromatographiques spécifiques d’une
famille d’énantiomères. Le facteur thermodynamique de la séparation
chirale a été ainsi mis en évidence.
Mais l’état actuel des différentes études, n’indique pas clairement s’il existe une corrélation entre la différenciation géométrique et la séparation physique. De nombreuses techniques utilisent pour la reconnaissance chirale la formation de complexes diastéréoisomères avec un complexant énantiomériquement pur. Une méthode telle que la Résonance Magnétique Nucléaire (RMN) permet de visualiser une reconnaissance chirale. En effet, les différents atomes (les protons, par exemple, qui sont facilement observables par RMN) des complexes diastéréoisomères ont des environnements magnétiques différents et présentent de ce fait des déplacements chimiques différents. Il y a reconnaissance chirale par différence géométrique. La chromatographie liquide haute performance (CLHP) permet de séparer physiquement des isomères par formation de complexes diastéréoisomères ayant des constantes de complexation différentes. Il y a reconnaissance chirale par différence thermodynamique.
La correspondance des résultats obtenus par les séparations chirales (thermodynamiques) et les discriminations chirales (géométriques) n’a jamais été formellement établie. Les diverses méthodes physiques d’analyse (RMN et CLHP plus particulièrement) utilisent des complexants différents. C’est pourquoi, il est apparu nécessaire d’étudier complètement un complexant sous ces deux modes de reconnaissance chirale.
Cette thèse a donc pour objet l’étude des relations entre les paramètres thermodynamiques et géométriques dans la reconnaissance chirale par des cyclodextrines modifiées.
Le choix du complexant chiral s’est porté sur les cyclodextrines car elles sont utilisées (sous des formes différentes) par les deux méthodes analytiques retenues, RMN et chromatographie liquide. Leur modification chimique a été effectuée car la compilation des études a démontré que la perte de la symétrie intrinsèque des cyclodextrines avait une influence positive sur leur capacité de reconnaissance chirale.
Les cyclodextrines permettent l’étude par RMN de discrimination chirale. Cette technique rapide et précise offre une évaluation de la reconnaissance chirale. En approfondissant celle-ci, l’accès aux géométries des différents complexes diastéréoisomères formés en solution est possible.
La réalisation de la séparation physique des isomères peut être facilement menée par chromatographie liquide. Cette technique physique, fournit un accès aisé au paramètre thermodynamique retenu pour caractériser les séparations (la constante de complexation de chacun des isomères vis-à-vis d’un même complexant). De plus, cette technique permet de conserver des conditions expérimentales proches de celles de la RMN.
Après l’étude des différentes facettes sous lesquelles la reconnaissance chirale a été étudiée, une problématique est apparue dans la difficulté de mettre en parallèle les résultats obtenus par séparation et par discrimination, ainsi que la difficulté d’homogénéisation des procédures d’études. Dans un premier temps, il semble nécessaire de mettre au point une famille de sélecteurs chiraux. Cette famille établie et caractérisée, l’étude du paramètre géométrique par RMN a été réalisée. L’étude thermodynamique a été effectuée par CLHP, avec le sélecteur chiral retenu, greffé sur colonne puis en solution dans l’éluant.
Cette thèse se divise donc en quatre grandes parties, tout d’abord le choix et le développement d’une famille de cyclodextrines, les 3,6-anhydro-b-cyclodextrines. Les synthèses et les caractérisations des dérivés di-anhydro seront décrites et suivies de l’étude du pouvoir complexant de ces molécules. Suite à ces premiers résultats, le choix du sélecteur chiral se portera sur la mono-3A,6A-anhydro-b-cyclodextrine,et, les synthèses ainsi que les analyses des dérivés di- et per-méthylés seront effectuées.
Une deuxième partie de ce mémoire portera sur l’utilisation de ces molécules complexantes, en RMN, pour déterminer leur pouvoir discriminant, sur différents composés racémiques.
La troisième partie portera sur l’étude par CLHP des couples cyclodextrine/racémique retenus suite aux résultats de la RMN. Les cyclodextrines seront dans un premier temps greffées sur colonne, puis, par souci d’homogénéiser les conditions d’analyses avec la RMN, l’étude chromatographique sera réalisée avec un sélecteur chiral dans l’éluant.
Enfin, la dernière partie sera un approfondissement de l’étude RMN. Les géométries fines des couples diastéréoisomères choisis seront étudiées pour tenter de mieux comprendre les différences observées entre RMN et CLHP.
1.La chiralité.
L’existence de symétrie et d’asymétrie remontent au début de l’univers mais la prise en compte de leur importance ne date que des deux derniers siècles. La découverte de leurs profondes implications dans la structure de la matière ainsi que dans les phénomènes de la vie est, elle, encore plus récente.
En 1883, soit plus de trente ans après avoir établi la dissymétrie moléculaire des cristaux, Pasteur1 affirme devant la Société chimique de Paris : "L’univers est un ensemble dissymétrique. Je suis porté à croire que la vie, telle qu’elle se manifeste à nous, doit être fonction de la dissymétrie de l’univers ou des conséquences qu’elle entraîne.[…] Tous les produits, pour ainsi dire, de l’œuf et de la graine sont asymétriques". De la simple molécule à l'organisme le plus complexe, rien de ce qui compose la vie ne reste semblable à lui-même de l'autre côté du miroir, et ce, sans qu’il soit actuellement possible d’en comprendre le pourquoi.
La chiralité est la faculté qu’à un objet à ne pas être identique à son image dans un miroir. L’exemple le plus facilement appréhendable d’objet chiraux, est celui de nos deux mains. Elles semblent identiques en tous points (nombre de doigts et organisation de ceux-ci), l’image de la main droite dans un miroir est la main gauche, mais elles ne sont pas superposables.
La plupart des organismes vivants présente, à des degrés divers, des caractères d’asymétrie dont le sens droit ou gauche est généralement bien défini. De tels caractères se retrouvent aussi bien dans la structure interne de la cellule des protozoaires, dans la morphologie des mollusques, dans l’enroulement de certaines plantes autour d’un tuteur, que dans l’arrangement interne des organes2.
Commençons par l'organisme le plus familier, le corps humain, chef-d’œuvre de la latéralisation : latéralisation anatomique (cœur à gauche, foie à droite), mais aussi latéralisation fonctionnelle. L’asymétrie de la nature va en effet bien au-delà de la morphologie puisqu’elle se manifeste également au niveau de notre cerveau. Cette asymétrie fonctionnelle du cerveau se traduit notamment par le fait que plus de 90% de la population se sert de la main droite pour exercer les activités les plus complexes (c’est d’ailleurs pour cela que nos tire-bouchons ont un pas de vis, une hélicité, droit).
D'autres exemples2, un peu moins proches : les plantes à enroulement hélicoïdal, tel le liseron (Convolvulus arvensis), ne grimpent autour de leur tuteur que dans un seul sens, la plupart suivent une spirale gauche. Mais quelques originales, le chèvrefeuille (Lonicera) par exemple, semblent avoir définitivement opté pour la spirale droite. De même chez les escargots, leur coquille forme généralement une hélice droite. Mais le buccin (famille du bulot et du bigorneau), parmi quelques autres, présente-lui un enroulement gauche. Là encore, il semble que la génétique fait loi. Parmi les espèces à coquille droite, les rares individus "gauchers" sont en effet porteurs d'une mutation spécifique.
De l'asymétrie des particules élémentaires à celle des êtres supérieurs, peut-on espérer trouver un lien de cause à effet ?
Lorsque l'on étudie les molécules de la vie on constate que toutes, ou presque, se présentent sous une seule des deux formes possibles. Ainsi les acides aminés, éléments constitutifs des protéines, n'existent pratiquement que sous forme lévogyre et, la connaissance de leur pourcentage dextrogyre permet la datation en géochronologie de composés organiques archéologiquement importants. Par contre la double hélice d'ADN répond au principe inverse et s’enroule naturellement vers la droite, bien que les synthèses actuelles permettent de créer des ADN à double hélice gauche.
Pourquoi la vie n'emploie-t-elle que des acides aminés de type gauche et des nucléotides de type droit ? Les protéines sont indispensables à la synthèse des molécules d'ADN (et inversement) on sait que la chiralité des unes a entraîné celle des autres. Mais, il semble que les premières molécules organiques apparues sur la Terre ne présentaient pas cette dissymétrie. Quand et pourquoi est apparue cette chiralité ?
A mesure que les
physiciens vont plus loin dans leurs connaissances des structures profondes
de la matière, ils découvrent que l'univers tout entier est asymétrique3.
Particules asymétriques, structures asymétriques, univers asymétrique,
l'origine de ces asymétries reste encore énigmatique.
Là, s'arrête la contribution de la physique à l'asymétrie du vivant. Car entre la chiralité des biomolécules et l'hélicité de la coquille d'escargot, il ne semble guère, cette fois, y avoir de relation directe. La latéralisation morphologique des plantes et des animaux trouvera sans doute son explication grâce aux avancées enregistrées dans un tout autre domaine, la génétique du développement. Une discipline dont les prémisses établis, il y a plus de quarante ans, sur la mouche Drosophila melanogaster, ont valu à Edward B. Lewis, Christiane Nüesllein-Volhard et Eric F. Wieschaus4 le prix Nobel de médecine 1995 (pour leur découverte sur le contrôle génétique du développement précoce de l’embryon).
Depuis une dizaine d'années, on découvre en effet que les grandes étapes de la morphogenèse sont dirigées par des "gènes du développement". Ceux-ci, véritables architectes moléculaires semblent universels et, contrôlent l'embryogenèse des êtres vivants. Les gènes qui déterminent l'axe antéro-postérieur et l'axe dorsol-ventral d’embryons d'animaux ont déjà été identifiés, et l'on est désormais en passe de recenser ceux qui gouvernent la latéralisation des organes.
En 1993, des chercheurs5 du Baylor College of Medicine de Houston (Texas) découvraient ainsi, chez la souris, que l'inactivation d'un seul gène entraînait la naissance d'individus présentant une inversion complète de l'emplacement de leurs organes. Ce gène, dont le dérèglement pourrait être responsable, chez l'homme, du situs inversus (affection qui atteint environ une personne sur 20 000 et qui se traduit par une inversion de l'emplacement des organes internes du thorax et de l'abdomen), pourrait être le premier d'une nouvelle famille de gènes du développement. De récents travaux6, menés sur le poulet, tendent en effet à démontrer que d'autres gènes, dès les premières quarante-huit heures du développement, s'expriment de manière différente d'un côté et de l'autre de l'embryon (gènes qui participeraient, entre autres, à la latéralisation du cœur).
Comme souvent lors des grandes avancées en science, l'épopée de la dissymétrie des molécules commence bien avant Pasteur7. Dès 1669, le physicien danois Erasmus Bartholin avait noté qu'un objet, regardé à travers le cristal transparent d'un minéral, le spath d'Islande (calcite CaCO3), produisait une double image. Il observa également, en faisant tourner le cristal, que l'une des deux images tournait avec lui, tandis que l'autre demeurait statique.
Il fallut attendre un siècle et demi pour comprendre ce phénomène de double réfraction, qui provient de la nature ondulatoire de la lumière. La lumière peut aussi être polarisée de manière à vibrer dans un seul plan.
Or les cristaux de spath d'Islande sont des polariseurs naturels qui n'acceptent que certains plans d'oscillation, en fonction de leur structure atomique.

Figure 1 : Représentation du phénomène de double réfaction de la lumière à travers le spath. Les deux rayons polarisés le sont de façon perpendiculaire à travers le cristal.
Eilhard Mitcherlich8 (1794-1863) découvre la loi de l'isomorphisme en 1819, et déclare "même cristal, même chimie". Il publie, en 1844, un article relatant le fait que les cristaux d'acide tartrique et d'acide racémique (mélange en proportions égales de l’acide tartrique sous ses deux formes chirales) sont isomorphes (même forme) mais l'acide tartrique est optiquement actif alors que l'acide racémique ne l'est pas !
En 1828, le physicien écossais William Nicol (1768-1851) utilisa le spath d'Islande pour fabriquer une paire de prismes. Leurs conceptions étaient ainsi faites que la lumière passant à travers eux puisse être soit avivée, soit obscurcie, selon la manière dont on faisait pivoter l'un des prismes par rapport à l'autre. Lorsqu'un composé organique en solution était placé entre ces deux prismes, l'un d'entre eux devait être tourné selon un certain angle pour permettre à la lumière toute entière de le traverser. Ainsi il devenait possible de mesurer de quel angle certaines solutions organiques font dévier le plan de polarisation de la lumière.
Jean-Baptiste Biot9 (1774-1862), poursuivant ces travaux, franchit l'étape qui devait permettre à Pasteur de découvrir la dissymétrie moléculaire. En 1815, Biot démontra que certaines substances organiques pouvaient faire dévier la lumière d'un côté ou de l'autre du plan de polarisation. Il supposa alors que ces changements arbitraires de direction provenaient d'une asymétrie des molécules elles-mêmes, mais il ne put aller plus loin.
2.Pasteur.
Louis Pasteur (1822-1895) avait une formation de chimiste et, c'est dans ce domaine qu'il fit ses découvertes les plus fondamentales. Celles-ci concernent particulièrement la dissymétrie des substances organiques.
En 1848, à vingt-six ans, Pasteur entreprend l'étude des tartrates, une famille de substances organiques qui exerce sur la lumière polarisée l'action observée par Biot. Pasteur pense que la différence d'activité optique correspond à une différence d'ordre moléculaire et il en fait son sujet de thèse10.
Sous son microscope, étudiant un par un les cristaux de tartrate, il découvre qu'ils ne sont pas tous identiques, mais se répartissent en deux groupes symétriques dans un miroir. Pourtant, l'assortiment de cristaux étudiés par Pasteur provient d'une solution qui, elle, ne fait par dévier la lumière polarisée. Pasteur soupçonne alors qu'il y a dans cette solution une quantité égale des deux éléments. Pour le vérifier, il sépare à la main, à l'aide d'une pince à épiler les deux types de cristaux. Puis il dissout chacun d'entre eux et mesure leur comportement vis-à-vis de la lumière polarisée à travers un prisme de Nicol. Comme il s'y attendait, une des solutions fait tourner la lumière dans un sens, la seconde dans l'autre11.
Pasteur confirma cette découverte sous le rigoureux contrôle de Biot et, forgea ainsi sa réputation. Dix ans plus tard, il démontra que les micro-organismes, entre deux formes optiquement actives d'une même substance, choisissent invariablement l'une d'entre elles pour leur nutrition ouvrant la voie de la maîtrise des cultures microbiennes. Effectivement Pasteur observe que la fermentation par Pénicillium glaucum du paratartrate ne s’effectue que sur l’isomère droit, le gauche n’est pas affectée par l’action des micro-organismes1.
Il trouve aussi différentes méthodes pour séparer les racémiques :
● faire des sels avec une amine active
● utiliser des micro-organismes.
Il existe de multiples façons de cristalliser les tartrates. Nonobstant, les cristalliser sous deux formes optiquement complémentaires, comme le fit Pasteur, relève de la chance autant que de l'habileté. Cette séparation ne s’effectuant qu’à une température précise de cristallisation de 27°C. Cependant, comme Pasteur le résuma lui-même dans une phrase devenue célèbre : "Dans les champs de l’observation, le hasard ne favorise que les esprits préparés."
Pasteur découvrit que de multiples molécules organiques existent sous leurs deux formes optiquement complémentaires. Mais ce fut le travail de ses successeurs, notamment Joseph Le Bel et Jacobus Van't Hoff, qui forgea le lien entre l’activité optique et la structure moléculaire.
3.Successeurs de Pasteur : Van’t Hoff et Le Bel.
L’existence du carbone asymétrique n’est reconnue que depuis 1874, grâce aux travaux de Le Bel et Van’t Hoff. En effet, Van’t Hoff12 (fondateur de la chimie physique et découvreur de la loi d'action de masse) et Jospeh Le Bel13 (1847-1930), proposent indépendamment la théorie du carbone asymétrique : dans une molécule, les liaisons du carbone sont orientées du centre du carbone vers les sommets d'un tétraèdre. Il n’y a d’activité optique que si les quatre groupes substituants sont différents.
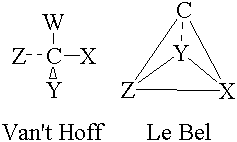
Figure 2 : Schéma représentant le carbone asymétrique d’après Van’t Hoff et Le Bel.
Le Bel avance une théorie sur l’asymétrie du carbone dans les composés optiquement actifs. Il publie ses idées indépendamment de Van’t Hoff et presque simultanément (Van’t Hoff semble avoir été influencé par Kekule, tandis que Le Bel considère le problème dans l’optique de Pasteur). Le Bel fut le premier à séparer un composé optiquement actif d’un mélange d’énantiomères. Il utilisa beaucoup, dans ce but, l’action sélective de certains micro-organismes sur des isomères dextrogyres ou lévogyres. Il fut aussi le premier à montrer que l’activité optique disparaît quand deux substituants du carbone sont identiques. Il a aussi prévu que d’autres éléments asymétriques pouvaient engendrer l’activité optique, ce qui fut vérifié expérimentalement par William Pope, en 1899. Ce qui conduit à la naissance de la stéréochimie.
Pasteur en 1848, parlait de molécules "dissymétriques" pour désigner ce qui est appelé aujourd’hui molécule chirale. C’est à partir du mot k h e i r (cheir, main) que lord Kelvin a forgé, à la fin du siècle dernier, le terme de chiralité pour désigner la propriété d’une figure géométrique de n’être pas superposable à son image.
Le caractère chiral, ou achiral, d’un objet est déterminé par la présence ou l’absence de certains éléments de symétrie. Les objets achiraux possèdent des éléments de symétrie inverse (plan, centre de symétrie). Les objets chiraux, qui ne peuvent posséder aucun de ces éléments, peuvent néanmoins présenter des axes de symétrie.
Les objets chiraux
peuvent ainsi exister sous deux formes, identiques par leurs dimensions et leurs
morphologies, et différentes par leurs géométries non superposables.
Il s’agit de formes énantiomorphes, souvent désignées comme
les mains par les adjectifs droit et gauche.
Cette propriété de chiralité est valable pour les objets microscopiques tridimensionnels que sont les molécules14. La chiralité moléculaire peut avoir des origines diverses : carbone asymétrique (symbolisé par C*) comme dans nombre de molécules organiques, absence d’éléments de symétrie (certains complexes de coordination, hélicènes par exemple).
Figure 3 : Gossypol . Dans les biaryles substitués en ortho de la liaison centrale la rotation des deux cycles benzéniques l’un par rapport à l’autre est bloquée. Ces molécules sont chirales si les deux groupes portés par chaque cycle sont différents. Le gossypol est présent dans les graines de coton, l’isomère (-) est un puissant contraceptif masculin15.
Les deux formes, images l’une de l’autre dans un miroir, des molécules chirales sont dites énantiomères (énantiomorphe étant plutôt réservé aux objets macroscopiques). On sait depuis Biot, que les deux énantiomères d’une molécule ont des effets opposés sur la lumière polarisée. L’énantiomère (+) ou dextrogyre dévie le plan de polarisation dans le sens des aiguille d’une montre, alors que le lévogyre (-) dans le sens opposé. La mesure de cette déviation est appelée le pouvoir rotatoire. Un mélange en quantité égale de ces deux isomères est qualifié de racémique et est inactif vis a vis de la lumière polarisée.
Le nom d’acide racémique (du latin racemus, raisin) a été donné par Gay-Lussac16 à un acide mystérieux, isolé en 1820 comme sous-produit du raffinage de l’acide tartrique (produit de la vinification), et présentant la même composition élémentaire que ce dernier bien qu’il soit dépourvu de pouvoir rotatoire. C’est Pasteur qui en 1848 a donné l’explication du phénomène : l’acide racémique est un mélange en quantité égale de l’acide (+) tartrique naturel et de son énantiomère (-) formé lors du raffinage sous l’effet de températures élevées17. Le terme racémique est maintenant universellement employé pour désigner les mélanges équimoléculaires d’énantiomères (+) et (-).
Ce qui distingue un énantiomère de son image c’est sa configuration absolue, soit la façon dont sont disposés les quatre substituants d’un carbone asymétrique. Un premier descripteur, l et d, de cette configuration a été introduit par Emil Fischer en 1891. Les lettres l et d utilisées par Fischer sont à différencier des l et d utilisées pour lévogyre et dextrogyre (changement de direction de rotation du plan de la lumière polarisée). Ce descripteur n’est plus très usité actuellement sauf pour ce qui concerne les acides aminés ainsi que certains sucres. Le système l/d est corrélé à la configuration chimique du carbone asymétrique du d-glyceraldehyde.
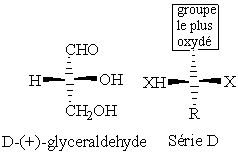
Figure 4 : Spécification de la configuration de Fisher ; la série l ou d à laquelle appartient la molécule est définie par la référence à une projection dans laquelle la chaîne la plus longue est disposée verticalement avec sa partie la plus oxydée vers le haut. Dans cette projection, les substituants disposés horizontalement sont en avant du plan, la chaîne étant en arrière.
E. Fisher reçu le prix Nobel en 1902 pour sa détermination de la configuration du (+)-glucose.
Ce descripteur a été supplanté par les R et S de Cahn, Ingold et Prelog dont l’usage introduit en 1966 est plus général. Il n’existe aucune relation directe entre la configuration absolue (R ou S) d’une molécule et le sens (+ ou -) de son pouvoir rotatoire.
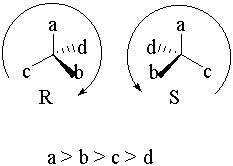
Figure 5 : Spécification de la configuration selon Cahn, Ingold, Prelog ; pour chaque carbone asymétrique de la molécule, on établit la séquence de priorité des substituants. Le C* est ensuite disposé de sorte que le groupe le moins prioritaire soit le plus éloigné de l’observateur. La disposition rictus ou sinister de la séquence a, b, c définit la configuration R ou S de ce C*.
Il existe aussi dans le cas des complexes octaédriques de Werner la désignation par les lettres D et L.
L D
C’est seulement dans les années 50 que les premières configurations absolues ont pu être déterminées par les méthodes expérimentales.
Chaque C*, chaque centre stéréogénique d’une molécule peut exister sous ces deux formes R et S. Ainsi, un C* unique donne deux molécules énantiomères, donc n C* conduiront à 2n stéréoisomères (au maximum) eux-mêmes constitués de 2n/2 paires d’énantiomères. Dans les cas du menthol, il y a trois C* (en position 1, 2 et 4 du cycle cyclohexanique), huit stéréoisomères sont prévisibles, répartis en quatre paires d’énantiomères : néoisomenthol, menthol, néomenthol et isomenthol.
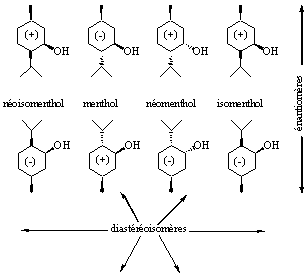
Figure 6 : Stéréoisomérie
dans la famille du menthol. Parmi les isomères de constitutions C10H20O,
le
3-p-menthol a été sélectionné, ses huit
stéréoisomères sont représentés.
Les stéréoisomères qui ne sont pas énantiomères sont appelés diastéréoisomères.
2. Chiralité et principes actifs.
Il aura fallu attendre la période récente et le révélateur qu’a constitué le drame de la thalidomide, pour que les conséquences de l’asymétrie - ou de la chiralité - du monde vivant soient prises en compte avec beaucoup plus d’attention. Jusqu’à ce drame, la chiralité des médicaments était peu prise en compte, dans la conception et la mise sur le marché de nouveaux médicaments.
La thalidomide, commercialisée en 1954 comme sédatif, non-barbiturique, possédant aussi une propriété calmante des nausées du premier trimestre de la grossesse, a provoqué dès 1956 la naissance de centaines d’enfants malformés. Il a été, par la suite, montré que seul l’énantiomère S-(-) de cette molécule était tératogène chez la souris. Cela ne signifie pourtant pas que l’administration du composé R-(+) seul aurait permis d’éviter ce drame. En effet, des études récentes ont montré que les énantiomères du composé se racémisent facilement à pH physiologique. De plus, différents métabolites de la thalidomide se révèlent être eux aussi tératogènes.
Une corrélation a été trouvée entre la date de prise et les malformations congénitales induites.
● 35-37ème jours : absence d’oreilles et surdité
● 39-41ème jours : absence de bras
● 43-44ème jours : phocomélie de trois doigts
● 46-48ème jours : pouce à trois phalanges
A ce jour, le mode d’action tératogène de la thalidomide n’est pas encore complètement élucidé et fait encore l’objet de nombreux débats passionnés.
Cependant, la thalidomide est de nouveau à l’étude pour son efficacité dans des cas de lèpre, d’aphtoses, de perte de poids liée au SIDA, ainsi que de réactions chroniques du greffon contre l’hôte. Toutefois son administration, qui a soulevé une importante vague d’émotion auprès des premières victimes, se fait en parallèle avec la prise de contraceptif.
Les stéréoisomères (énantiomères comme diastéréoisomères) ont généralement un comportement et des effets biologiques différents. En effet, l’action d’un médicament est l’aboutissement d’une séquence complexe dans laquelle on peut distinguer généralement trois phases principales consécutives. 1) Une phase d’exposition qui permet d’introduire dans l’organisme par inhalation, injection, digestion ou contact cutané, la dose de principe actif. 2) Une phase pharmacocinétique, au cours de laquelle la molécule active va être la cible de protéines qui vont la transporter, la transformer en métabolites (pouvant être eux-mêmes pharmacologiquement actifs) ou la détruire. 3) Une phase pharmacodynamique où ce qui restera de la molécule initiale (ou de ses métabolites) va se fixer sur les récepteurs moléculaires pour produire les effets recherchés (ou non). Si la molécule active est chirale, chacune de ces étapes mettant en jeu des interactions avec les biopolymères chiraux sera stéréosélective.
On appelle agoniste la molécule exerçant l’activité principale, et antagoniste celle sont l’action s’oppose à l’activité principale.
Les relations entre les activités biologiques de chaque stéréoisomère pur et celles de leurs mélanges sont souvent très complexes et, leur analyse nécessite des études très détaillées. Quelques situations typiques peuvent toutefois être distinguées comme il sera décrit dans la suite.
Il arrive que deux énantiomères aient des activités similaires : c’est le cas de nombres d’anesthésiques locaux tels la bupivacaïne, dont l’énantiomère (-) est cependant légèrement plus actif que l’énantiomère (+) en raison de son pouvoir vasoconstricteur. L’utilisation de tels composés sous forme racémique ne présente donc aucun inconvénient.
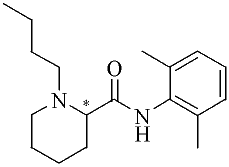
Figure 7 : Bupivacaïne18 - chlorhydrate de butyl-1 diméthyl-2',6' pipéridinecarboxanilide-2. Elle agit au niveau du neurone en interférant avec le processus d'excitation et de conduction.
Même lorsque les deux énantiomères ont une même activité, il peut se produire que l’un d’entre eux soit métabolisé plus rapidement et conduire à des produits toxiques ; c’est le cas de la prilocaïne, autre anesthésique local. L’isomère (-) est rapidement métabolisé par un mécanisme hépatique dans l’organisme en ortho-toluidine hautement toxique (oxydation de l’hémoglobine).
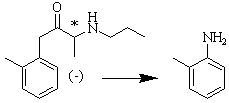
Figure 8 : La (-) prilocaïne19 (chlorhydrate de alpha-propylamino-2-méthylpropionanilide se métabolise en ortho-toluidine. La prilocaïne présente le même mode d’action que la bupivacaïne.
Dans de multiples cas, seul un énantiomère présente l’activité recherchée, le second étant inactif et dépourvu d’activités non désirées à la dose administrée. Dans ce cas, l’utilisation du principe actif sous forme racémique peut être justifiable, mais la commercialisation d’un énantiomère pur semble nettement préférable dans le cas de doses thérapeutiques élevées. L’intérêt est double. Tout d’abord, le patient trouvera certainement plus confortable de ne prendre qu’un ou deux grammes de produits plutôt que le double, et le fabricant aura à synthétiser moitié moins de principe actif. C’est le cas du naproxène, anti-inflammatoire non stéroïdien (AINS) dont seul l’isomère S-(+) est produit. Le fait de commercialiser un énantiomère pur semble devenir un argument de marketing, c’est aussi une technique qui permet de remettre sur le marché d’anciennes molécules en améliorant leur efficacité (dose et effets secondaires moindres).
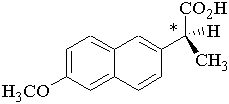
Figure 9 : S-(+)-naproxène20 (acide(+)-(methoxy-6 naphtalène-2)-2 propionique) Il agit par inhibition de la cyclo-oxygénase et de la synthèse des prostaglandines.
Les deux énantiomères ont une constante de complexation semblable vis à vis d’un même récepteur mais leurs actions sont opposées, l’un est agoniste, l’autre antagoniste. Une variante de ce cas est celui où les deux énantiomères ont des effets opposés sans être en compétition (ils agissent sur différents récepteurs). C’est le cas de certains barbituriques pour lesquels l’isomère (-) est dépresseur alors que l’isomère (+) provoque des convulsions. Ces circonstances ne sont évidemment pas favorables à l’utilisation du produit sous forme de racémique.
L’énantiomère inactif vis-à-vis de l’effet recherché, inhibe un effet secondaire nocif de l’isomère actif (ou d’un métabolite). C’est le cas de certains diurétique de la famille de l’indacrinone. L’énantiomère (+) de ce principe actif est diurétique mais entraîne une rétention d’acide urique. L’isomère (-), lui favorise l’élimination de cet acide urique. Dans ce cas, l’administration du produit sous forme racémique peut sembler préférable. Mais, la composition 1:1 n’est pas obligatoirement celle présentant le meilleur effet thérapeutique. Dans le cas du diurétique cité, il a été démontré que le rapport 1:8 des deux énantiomères semble être optimum.
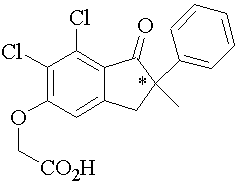
Figure 10 : Indacrinone21 (MK-196) l’indacrinone bloque les canaux Cl, elle n’est commercialisé qu’aux USA. L’indacrinone est un diurétique de l’anse de la famille de l’acide ethacrynic.
Un des énantiomères possède l’activité désirée mais l’autre est source d’effets secondaires. Il existe de nombreux exemples de cette situation (dont la thalidomide précédemment citée). Dans le cas de l’éthambutol, composé antituberculeux, seul l’isomère de configuration S,S-(+) est tuberculostatique tandis que le R,R-(-) occasionne la cécité.
Figure 11 : S, S-(+)-éthambutol22. Il agit sur les bacilles tuberculeux en phase de multiplication, il réduirait leur biosynthèse de l'ARN et de l'ADN. L’isomère toxique entraîne une névrite optique due à une déplétion en zinc.
Pour ce qui est de la pénicillamine, l’énantiomère d-(-) est un antirhumatismal, tandis que l’isomère l-(+), dépourvu de cette activité, est hautement toxique et est suspecté d’être cancérigène.
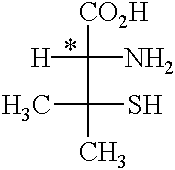
Figure 12 : Pénicillamine23 (3-mercapto-d-valine). Chélateur du plomb, du cuivre, du mercure, de l’or et du zinc, anti-inflammatoire.
Dans de telles situations, l’utilisation de l’énantiomère actif sous une forme optiquement pur est obligatoire. De plus, il est indispensable de vérifier que cet isomère ne subit pas une inversion de configuration dans l’organisme (comme la thalidomide).
Il existe aussi le
cas où les deux énantiomères ont séparément
et à des doses semblables des activités différentes et
utiles. C’est par exemple le cas du couple
2R,3R-(+)-dextropropoxyphène et de son énantiomère 2S,3S-(-)-lévopropoxyphène.
Le premier isomère est commercialisé sous le nom de Darvon, il
est analgésique. Le second, qui est commercialisé sous le nom
de Novrad est antitussif. Il est amusant de noter que ces deux produits ont
été commercialisés aux USA sous des noms "énantiomères"
(palindromes).
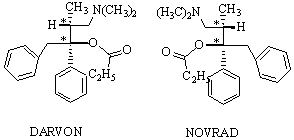
Figure 13 : Darvon24 : chlorhydrate de (+)-propionate de benzyle-1 diméthylamino-3 méthyle-2 phényle-1 propyle. Action analgésique de type morphine; agoniste des récepteurs m., il bloque les synapses dans le cheminement central de la douleur. Il présente une activité similaire à celle de la méthadone (mais est 1/8 fois moins actif).
Bien qu’il ne s’agisse pas de médicaments, citons aussi le cas du menthol qui est utilisé en parfumerie, en cosmétique et dans l’industrie alimentaire exclusivement sous la forme de son énantiomère (-) (celui présent dans les feuilles de menthe). La raison en est que l’énantiomère (+), même s’il possède une odeur mentholée, ne présente pas du tout le caractère piquant et rafraîchissant qui fait le succès du produit image, il a en effet, l’odeur des graines de carvi (Carum carvi).
Citons aussi le cas du limonène : l’isomère R-(+) est présent dans l’orange alors que l’isomère S-(-) lui se retrouve dans le citron : configurations différentes, odeurs différentes.
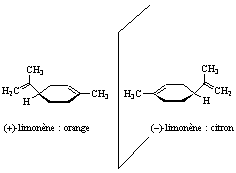
Figure 14 : Des isomères à odeurs très différentes25.
Comme il a été
détaillé précédemment, les isomères optiques
biologiquement actifs, peuvent présenter une très large diversité
dans leurs actions (complémentaires ou non).
Il apparaît donc indispensable de procéder à des études
biologiques sur un produit optiquement pur afin d’évaluer ses activités/toxicités
propres (ainsi que ses métabolites).
Pour pouvoir procéder à de telles études pharmaceutiques,
le chimiste doit obtenir les composés de façon optiquement pure
(directement après synthèse, ou procéder à leur
séparation).
3. Obtention d’isomères optiquement purs.
La production d’isomères optiquement purs destinés à être utilisés comme médicaments reste un des défis majeurs de l’industrie pharmaceutique. La tendance à la production systématique de médicaments énantiomériquement purs semble bien amorcée, tendance qui se trouve renforcée par les nouvelles directives de la pharmacopée européenne.
En 1990, le marché mondial
des médicaments était constitué de 43% de composés
achiraux, 25% de racémiques, 28% d’énantiomères purs d’origine
naturelle ou
semi-synthétiques, et seulement 3% d’énantiomères produits
par synthèse. En 1995, dans le portfolio des produits en développement
chez Eli Lilly & Co, ces chiffres deviennent respectivement 23%, 0%, 23%
et 54% soit 77% pour les énantiomères purs et aucun racémique.
Pour la préparation d’énantiomères purs, les industriels disposent de différentes méthodes.
1. Synthèse biologique ou quand la nature forme le C*.
La première méthode utilisée consiste en l’utilisation comme matière première de produits énantiomériquement purs, disponibles en grande quantité (et à bas prix). Ces produits optiquement purs proviennent de sources naturelles (généralement végétales) ou industrielles. Ainsi le d-glucose, l’une des substances chirales les plus abondantes, est la matière première de la synthèse industrielle de l’acide l-ascorbique. Cet acide est lui-même utilisé comme point de départ pour la synthèse de certains médicaments de la famille des b-bloquants.
La culture de plantes spécifiques, source de molécules chirales complexes, est très usitée. Ces molécules, optiquement pures, de départ sont ensuite transformées par hémisynthèse en principes actifs de façon relativement simple et surtout non racémisante pour les carbones asymétriques.
2. Synthèse asymétrique ou quand le chimiste imite la nature.
La synthèse asymétrique consiste à fabriquer une substance chirale sous une forme non racémique (idéalement sous la forme d’un seul énantiomère) en utilisant comme point de départ une molécule achirale. Dans ce cas, la chiralité est introduite par le biais d’un catalyseur chiral. La l-dopa (antiparkinsonien) et le (-)-menthol sont produits industriellement par synthèse asymétrique.
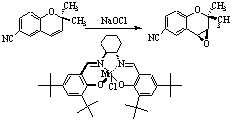
Figure 15 : Préparation d’un intermédiaire pharmaceutique énantiomériquement pur par synthèse asymétrique. Les deux faces de la double liaison sont énantiotopiques. Le catalyseur chiral favorise l’époxydation sur une face préférentiellement.
La synthèse asymétrique a considérablement progressé au cours des vingt dernières années notamment sous l’influence de H. Kagan.
Une variante de la synthèse asymétrique est d’utiliser des catalyseurs biologiques (des enzymes) pour effectuer certaines réactions. La production industrielle d’acide l-aspartique par réaction de l’acide fumarique avec l’ammoniaque en présence de l’aspartase en est un exemple.
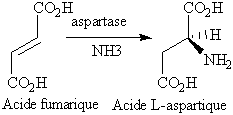
Figure 16 : Production industrielle
de l’acide l-aspartique par synthèse asymétrique enzymatique.
Même principe que pour l'époxydation asymétrique.
L’enzyme sélectionne l’une des faces énantiotopiques de
l’acide fumarique pour y additionner la molécule d’ammoniaque.
Actuellement, le dédoublement des racémiques obtenus par synthèse classique représente toujours l’une des principales voies d’accès aux composés énantiomériquement purs. Aujourd’hui près des deux tiers des molécules actives chirales issues de synthèse proviennent de dédoublements. Il existe différentes possibilités pour séparer physiquement des isomères.
1. La dérivatisation.
La dérivatisation consiste à combiner le racémique à dédoubler (± )-A avec une substance auxiliaire elle-même énantiomériquement pure, (+)-B. La combinaison se fait par liaison covalente entre les deux espèces. On obtient ainsi un mélange de deux composés diastéréoisomères, [(+)-A,(+)-B] et [(-)-A,(+)-B]. Ces diastéréoisomères présentent des caractéristiques physico-chimiques différentes et peuvent donc ainsi être séparés par cristallisation ou par chromatographie. Après séparation des diastéréoisomères, suivie du clivage et de la récupération de l’auxiliaire B, on obtient ainsi les deux énantiomères de A de façon optiquement pure. Le principal intérêt de la dérivatisation est que cela permet l’utilisation de colonnes chromatographiques conventionnelles.
Les méthodes de séparation chirale nécessitant une étape utilisant un réactif optiquement pur ont néanmoins plusieurs limitations et peuvent fournir un résultat erroné quant à la composition énantiomérique. En effet, une racémisation lors de la réaction, l’incertitude quant à la pureté optique du réactif26, et une discrimination chirale de la réactivité de B peuvent affecter de façon importante la détermination quantitative de l’excès énantiomérique. La dérivatisation par un réactif achiral peut être aussi utilisée limitant ainsi certaines des difficultés occurantes lors de la réaction27.
Il existe une variante biologique du dédoublement, dans laquelle sous l’action d’un système enzymatique ou d’un micro-organisme, l’un des isomères est transformé en produit facilement séparable de l’autre énantiomère. Il est facile de comprendre la limitation de ce procédé par la difficulté à trouver, quand il existe, le composé biochimique idoine.
2. Cristallisation.
Le dédoublement par cristallisation directe du racémique (ou dédoublement par entraînement) peut s’effectuer dans les cas où celui-ci est formé de cristaux dont les énantiomères se sont séparés spontanément. Ce type de racémique, appelé conglomérat, est relativement peu fréquent, c’est celui rencontré par Pasteur. Il existe cependant plusieurs applications industrielles, notamment pour la production de (-)-menthol (le benzoate de menthyle est un conglomérat), et de la l-a-méthyldopa (principe actif antihypertenseur).
Mais le dédoublement par cristallisation de diastéréoisomères reste de loin la méthode la plus employée dans l’industrie. Deux des exemples les plus spectaculaires de ce type de procédé sont les dédoublements de la phénylglycine et du naproxen qui ont en commun le fait que l’énantiomère non désiré est racémisé et recyclé, de sorte que 100% du racémique se trouve être transformé en l’énantiomère désiré.
3. Méthodes chromatographiques préparatives.
Plusieurs méthodes chromatographiques sont utilisées pour la séparation chirale d’une large variété de composés. La résolution directe d’énantiomères, soit par l’utilisation d’une colonne à phase stationnaire chirale, soit par l’utilisation d’additifs chiraux dans la phase mobile, est généralement une voie plus élégante de séparation des énantiomères que par synthèse de composés diastéréoisomères à l’aide d’un réactif optiquement pur.
Des techniques directes de séparation d’énantiomères sont basés sur la formation de diastéréoisomères par liaisons de Van der Waals (complexation) entre le mélange que l'on veut séparer et un isomère pur. La formation réversible de ces complexes diastéréoisomères permet une séparation complète et une détermination quantitative de chaque énantiomère, même si le sélecteur chiral n’est pas optiquement parfaitement pur28.
Ces dernières années de nombreuses colonnes chirales sont apparues sur le marché, utilisant différentes catégories de phases chirales. Il en existe une très large variété : de type protéines-enzymes (albumine de sérum humain, greffage de a-chymotrypsine par exemple), Pirkle (complexes p/liaisons H), polymères (esters cellulosiques, carbamates cellulosiques, carbamates amylosiques), échanges de ligands, inclusion (cyclodextrine, éthers couronnes), transfert de charge. Quant aux mécanismes de reconnaissance chirale, ils sont très nombreux (donneur/accepteur d'électrons p pour les sélecteurs de Pirkle, complexation chirale pour les sites donneurs ou accepteurs de protons, complexes d'inclusion pour les macromolécules).
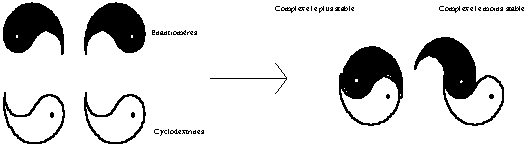
Figure 17 : Mécanisme de la séparation chirale
par formation de complexes diastéréoisomères
d’après Pirkle29 (en blanc l’isomère
chiral pur et en noir celui à séparer).
Les sélecteurs chiraux antibiotiques connaissent actuellement un important développement, notamment ceux à base de teicoplanine et de vancomycine, que se soit par CCM30, par CLHP (phase inverse ou directe)31, ou par électrophorèse capillaire. De même, les sélecteurs chiraux à bases cyclodextrines connaissent un important développement grâce à la richesse de taille de cette famille de sélecteurs, permettant ainsi leur utilisation pour une large diversité d’énantiomères.
Il est à noter que des séparations chirales ont put être obtenues à partir d’éthers couronnes non chiraux32.
Quelles que soient les méthodes d’obtention des composés optiquement purs, il faut pouvoir effectuer un contrôle de la pureté optique.
1. Le pouvoir rotatoire.
Le procédé le plus ancien de vérification de pureté optique, qui comme explicité précédemment est à l’origine de la découverte de la chiralité, est l’utilisation des pouvoirs rotatoires opposés des énantiomères. Le pouvoir rotatoire spécifique d’un énantiomère fait partie des caractéristiques physico-chimiques de celui-ci et est l’objet de nombreuses tables de références. L’angle de rotation de la lumière polarisée, induit par une solution d’énantiomère à contrôler, est mesuré. Cette mesure est effectuée en degré et ramenée à la valeur référence a pour une concentration du produit de 1g/cc dans une cellule de 1dm de long à une longueur d’onde l. Les énantiomères ayant des pouvoirs rotatoires d’une même valeur absolue mais de signe opposé, la mesure du pouvoir rotatoire du produit à analyser permet le calcul de sa pureté optique (ou excès énantiomérique).
|
Pourcentage de pureté optique = |
Pouvoir rotatoire du mélange |
* 100 |
|
Pouvoir rotatoire spécifique de l’énantiomère |
Malheureusement, la polarimétrie est peu sensible33 et ne permet pas une certitude quand à une totale pureté optique d’un composé. De plus cette vérification nécessite une connaissance préalable de la valeur du pouvoir rotatoire pour le composé à étudier ce qui en limite l’utilisation. Le dichroïsme circulaire est bien plus sensible que la polarimétrie, mais nécessite l’existence d’un groupement chromophore en UV pour la détection du signal8.
2. La chromatographie.
Cette méthode utilisée sous une forme analytique peut permettre le contrôle de la pureté optique d’isomères. Le principe de base, à savoir la formation de composés diastéréoisomères par complexation reste identique. Dans le cadre de contrôle analytique, les méthodes chromatographiques utilisées peuvent être différentes : électrophorèse capillaire34, chromatographie sur couches minces (CCM), chromatographie gazeuse (CG)35.
La chromatographie gazeuse connaît un renouveau depuis la conception de colonnes capillaires stables thermiquement. Ce regain d’intérêt est particulièrement développé pour la séparation d’énantiomères, petits, ou non aromatiques, fréquemment utilisés en synthèse asymétrique. De plus, la grande résolution des colonnes capillaires en CG, rend cette méthode idéale pour l’analyse de composés complexes d’origine biologique (arômes par exemple). Néanmoins, cette technique connaît différentes limites : les composés à analyser doivent être volatils, la température élevée à laquelle s’effectue l’analyse peut accélérer la racémisation de la phase stationnaire ou de l’analyte, de même cette température diminue les différences de constantes de complexation des énantiomères pour la phase stationnaire.
3. La RMN.
La RMN n’est d’utilisation que fort récente quant à la détermination de pureté optique36. Deux énantiomères présentent un spectre identique en RMN mais, les composés diastéréoisomères ont des spectres RMN différents. Il est donc apparu, que la formation de complexes diastéréoisomères, obtenus par l’adjonction d’un complexant chiral aux isomères étudiés, peut entraîner un dédoublement de spectre, même partiel. Cette méthode est relativement sensible (détection d’une impureté de 1%) mais connaît aussi des limites. Il faut que le dédoublement de pics quand il advient, soit observable et non dissimulé par le signal du complexant par exemple. De même ce dédoublement n’intervient pas systématiquement ou n’est pas suffisamment important pour que deux pics distincts, même partiellement, puissent être observés.
4. Choix d’un complexant : les cyclodextrines.
Il existe un large choix de molécules complexantes chirales, le choix pour cette étude s’est porté sur les cyclodextrines.
Les cyclodextrines
font sûrement parties des premières molécules macrocycliques
étudiées pour leurs capacités à former des composés
d’associations. Ces oligosaccharides cycliques naturels ont participé
à l’ouverture de la voie de la chimie supramoléculaire, domaine
de recherche particulièrement fécond, à l’interface de
la synthèse organique
(ou minérale), de la physico-chimie et de la biologie et, qui a littéralement
explosé durant les trois dernières décennies.
En 1891, Villiers37 isolait pour la première fois un groupe d’oligosaccharides non réducteurs provenant de la dégradation enzymatique de l'amidon par une amylase (cyclodextrine glucosyl transférase) produite par différents bacilles dont Bacillus macerans. Villiers a isolé 3 g d’une substance cristalline ((C6H10O5)2 3H2O) à partir de la digestion de 1 kg d’amidon par une souche de micro-organismes. Ces produits (Villiers met en évidence la présence de deux produits, probablement l’a- et la b-cyclodextrine) possèdent des particularités physico-chimiques proches de celles de la cellulose, il les baptise donc "cellulosines".
Schardinger38,
20 ans plus tard, isole la souche microbienne responsable de la formation de
ces "cellulosines", qu’il dénomme Bacillus Macerans39
et, décrit le mode de purification et de préparation de ces oligosaccharides.
Il met aussi en lumière la capacité de ces dextrines (appellation
générale des produits de dégradation de l’amidon) à
former des adduits particuliers avec les molécules de diiode. La distinction
entre l’a-dextrine
et la b-dextrine
est due à leur différence quant aux complexes cristallins formés
avec l’iode. Le complexe
de l’a-dextrine
est gris-vert alors que celui formé par la b-dextrine
est
rouge-brun.
C’est en 1932 que Prigsheim40 et son équipe démontrent que ces produits ont la propriété de former des complexes avec des molécules organiques.
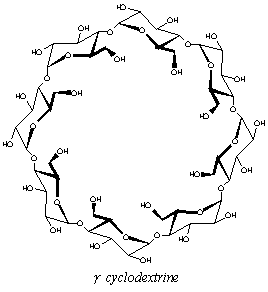
D. French, F. Cramer et K. Freudenberg contribuèrent également grandement à la connaissance des cyclodextrines et à l'élucidation de leur structure durant les années 30-40. Freudenberg et son équipe démontrent alors que ces oligosaccharides sont constitués d’un enchaînement de n unités a-d-glucopyranosidiques, la fraction principale contenant l’alpha- et la bêta-cyclodextrine (possédant respectivement 6 et 7 unités). De même le postulat en la capacité des cyclodextrines à former des composés d'inclusion41 est posé. C’est cette même équipe qui, en 1948, découvre la g-cyclodextrine (constituée de 8 unités glucose) et qui détermine entièrement sa structure42.
Au début des années 1950, les équipes de French et de Cramer étudièrent de façon intensives les productions enzymatiques de cyclodextrines, leurs purifications, et leurs caractérisations physico-chimiques.
La structure de la d-cyclodextrine a été publiée en 199043.
 |
 |
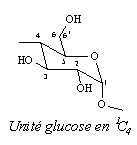 |
Figure 18: Formule générale des CDs et représentation schématique de leur structure tridimensionnelle.
Cette propriété des cyclodextrines à former des complexes d’inclusion devient alors le sujet d'études intensives, notamment par l'équipe de Cramer44. C'est ainsi que le tout premier brevet est déposé concernant l’application des cyclodextrines pour la mise en forme d’un composé à activité biologique en 195345. A partir de ce moment, on observe une recrudescence de l'étude des cyclodextrines, tant du point de vue de leur fabrication industrielle, que de l’exploitation de leurs propriétés, de leurs modifications chimiques ou bien encore, de leurs domaines d’applications46.
Les cyclodextrines (CDs) sont donc des oligosaccharides cycliques non-réducteurs obtenus industriellement par dégradation enzymatique de l’amylose (forme linéaire de l’amidon) à l'aide d’une enzyme, la cyclodextrine glucosyltranférase (CGTase). Les trois types de CDs les plus couramment rencontrés sont l’a-, la b- et la g-CD, qui sont constitués respectivement de 6, 7 et 8 unités d-glucopyranosiques, liées en a-1,4.
1. Nomenclature.
L’appellation IUPAC de la b-cyclodextrine, est : 5,10,15,20,25,30,35 -heptakis(hydroxymethyl)-2,4,7,9,12,14,17,19,22,24,27,29,32,34-tétradécaoxaoctacyclo [31.2.2.23,6.28,11.213,16.218,21.223,26.228,31] nonatétracontane-36,37,38,39, 40,41,42,43,44,45,46, 47,48,49-tétradécole. Dans un souci de clarté, on retrouve diverses appellations dans la littérature : b-dextrine de Schardinger, cyclomaltoheptaose, cycloheptaglucane, cycloheptaamylose, b-CD, ou bien encore C7A pour la b-cyclodextrine. Dans cette étude le terme b-CD ou b-cyclodextrine sera utilisé.
2. Structure géométrique et propriétés.
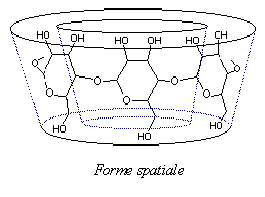
Les structures tridimensionnelles des CDs ont pu être obtenues à partir de l’étude de leur monocristaux par diffraction des rayons X (et même, de quelques monocristaux de complexes CD-invitée) ce qui a permis de mettre en évidence la structure tronconique des CDs ainsi que de déterminer les dimensions des cavités de chacune d’elles47.
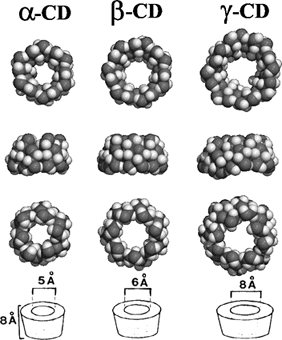
Figure 19: Structures tridimensionnelles des cyclodextrines naturelles (a-, b-, et g-CD de gauche à droite), avec de haut en bas: une vue de la face des hydroxyles secondaires ("grand côté"), une vue latérale et, une vue de la face des hydroxyles primaires ("petit côté"). En bas, les dimensions respectives des CDs obtenues d’après les données cristallographiques.
Ces études ont permis de montrer que l’extérieur des CDs est tapissé par les fonctions hydroxyles des unités glucose, tandis que les atomes de carbone et d’hydrogène tapissent l’intérieur de la cavité. La structure des cyclodextrines, alliée à l'orientation particulière adoptée par les diverses fonctions hydroxyles des unités glucopyrannose, donnent aux CDs leurs caractères amphiphiles caractéristiques, dût à un extérieur relativement hydrophile (surface de contact avec le solvant) et un cœur relativement hydrophobe (surface de contact avec la molécule invitée).
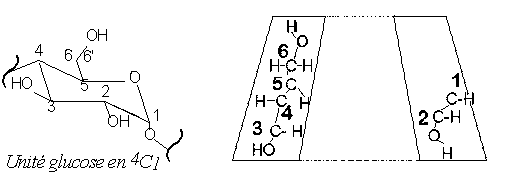
Figure 20: Représentation d’un cycle a-d-glucose en conformation chaise 4C1 et représentation schématique de la coupe transversale d’une cyclodextrine avec numérotation des atomes de carbones.
Les cyclodextrines présentent donc une forme torique, le côté le plus étroit étant appelé face primaire (les hydroxyles primaires y étant situés) et le côté le plus large, face secondaire (les deux groupes hydroxyles secondaires de chaque unité y étant localisés). De plus, on entrevoit sur la figure, que les hydroxyles primaires et secondaires forment un réseau dense de liaisons hydrogène, contribuant ainsi à la rigidité du macrocycle, et stabilisant la forme tronc-conique des molécules. Cette structure spatiale des CDs est aussi responsable de certaines de leurs caractéristiques physico-chimiques.
La cavité interne du tore est relativement apolaire car tapissée de deux couronnes de groupes CH (protons H3 près de la face secondaire et protons H5 près de la face primaire), séparées par les oxygènes glucosidiques. On peut distinguer sur la figure ci-dessus que tous les protons H3 et H5 des différentes unités glucose pointent vers l’intérieur de la cavité des CDs, particularité importante pour l’étude par Résonance Magnétique Nucléaire (RMN) des propriétés d’inclusion de ces molécules, comme cela sera explicité par la suite.
Il est important pour la suite de notre étude de noter que les différentes unités constitutives des CDs se trouvent dans la conformation chaise 4C1, c'est à dire que le carbone 4 se trouve au-dessus du plan moyen du cycle glucosidique, alors que le carbone 1 se trouve en dessous de ce plan moyen.
Tableau 1: Caractéristiques physico-chimiques des principales cyclodextrines48
|
a-CD |
b-CD |
g-CD |
|
|
Nombre d’unités glucose |
6 |
7 |
8 |
|
M (g/mol) |
972 |
1135 |
1297 |
|
Formule brute |
C36H60O30 |
C42H70O35 |
C48H80O40 |
|
Solubilité dans l’eau à 298K (g/L) |
145 |
18,5 |
232 |
|
[a]d 25°C |
150 ± 0,5 |
162,5 ± 0,5 |
177,4 ± 0,5 |
|
Volume approximatif de la cavité (106 pm3) |
174 |
262 |
427 |
|
Diamètre de la cavité (grand côté-petit côté) ( ) |
4,7-5,3 |
6,0-6,5 |
7,5-8,3 |
Il est à remarquer la faible solubilité de la b-CD en comparaison de celles de l’a- et de la g-CD. Cette perte de solubilité, dont les causes n’ont pas été totalement éclaircies, semble due au réseau de liaison hydrogène particulièrement fort dans le cas de CD à 7 unités. Lors de synthèses de b-CDs modifiées, mono- ou poly-modifications, les solubilités obtenues sont alors très largement augmentées par rapport à la CD naturelle, y compris après greffage de groupements relativement hydrophobes, renforçant l’hypothèse du réseau stabilisant. Dans le cas de l’a-CD cette ceinture de liaisons H est incomplète, l’une des unités étant dans une position distordue, il n’y a donc que 4 liaisons formées (au lieu des 6 prévues)49.
Pour ce qui est des CDs de taille supérieure50 d-, e-… (et de la g-CD dans une moindre mesure) celles-ci se présentent sous forme de cylindre non régulier, effondré en leur centre, de ce fait, leurs cavités se trouvent être plus petite que celle de la g-CD.
1. Inclusion.
L'intérêt des cyclodextrines provient essentiellement du caractère hydrophobe de leur cavité, la face extérieure étant hydrophile. Les CDs sont, de ce fait, des composés de choix pour l'inclusion de molécules hydrophobes à la condition que la taille de ces invités s'accorde aux dimensions internes de la cavité. Les CDs peuvent ainsi inclure partiellement ou en totalité un composé invité, ce qui donne alors lieu à la formation de complexes comportant éventuellement plusieurs molécules (1 ou 2) de CD ou de molécules invitées. De nombreux exemples de complexes CDs-invité, avec divers arrangements structuraux, se trouvent ainsi décrits dans la littérature. Les structures des principaux complexes ayant fait l’objet de publications sont représentées sur la figure suivante.
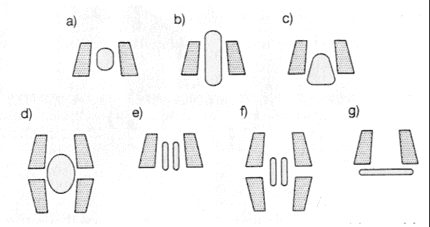
Figure 21: Diverses structures de complexes cyclodextrines-invités en solution aqueuse décrits dans la littérature a) inclusion complète ; b) inclusion "axiale" ; c) inclusion partielle ; d) complexe 2:1 ; e) complexe 1:2 ; f) complexe 2:2 ; g) complexe "non-spécifique".
Ces structures peuvent
être mises en évidence et caractérisées en employant
diverses techniques d'analyses physico-chimiques, comme la spectroscopie UV-visible,
la spectroscopie de fluorescence, l'analyse cristallographique, la spectroscopie
RMN ou bien encore, à l'aide des méthodes d'analyses électrochimiques51.
La taille des CDs peut être un facteur limitatif important quant à leur capacité à former des complexes d’inclusion et à leurs stabilités relatives. Ainsi, en relevant diverses valeurs de constantes de complexation (K de l'équilibre hôte-invité, dans des cas de complexes 1:1) parues dans la littérature, Connors a tracé la fréquence de distribution des constantes de complexation pour les complexes formés par l’a-, la b- et la g-CD52.
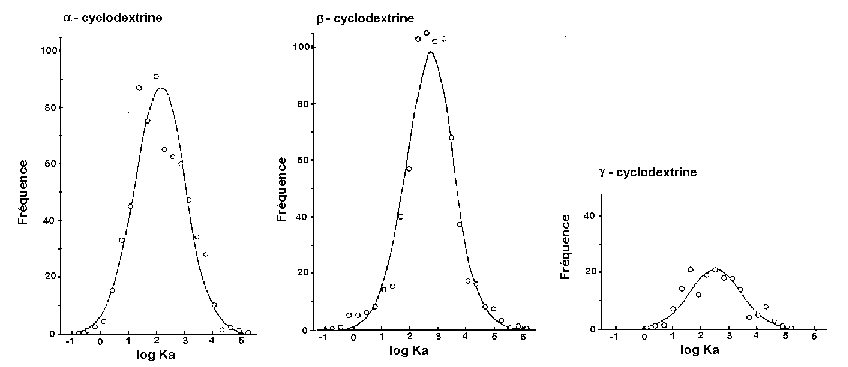
Figure 22: Distribution des constantes de complexation des complexes 1:1 formés par les différentes cyclodextrines naturelles.
Une distribution quasi-gaussienne de ces constantes de complexation est observée, quelle que soit la CD naturelle considérée. Une valeur moyenne de K a pu alors être extraite pour chacune de ces CDs, dans le cas de complexes 1:1 (avec s l’ecart type observé).
Tableau 2: Paramètres utilisés pour établir les courbes de distributions des constantes de complexation des différentes CDs dans le cas de complexes 1:1.
|
Molécule hôte |
Nombre de constantes de complexation prises en compte |
Nombre de systèmes hôte-invité 1:1 différents |
log (K)moy. (M-1) |
s (M-1) |
|
a-CD |
960 |
663 |
2,11 |
0,90 |
|
b-CD |
1142 |
721 |
2,69 |
0,89 |
|
g-CD |
188 |
166 |
2,55 |
0,93 |
Il apparaît alors, que toutes les cyclodextrines naturelles présentent une valeur moyenne de constante de complexation relativement faible (Ka~103 M-1), valeur ne présentant pas l’incompatibilité avec leur utilisation comme sélecteur chiral en chromatographie. Il est à noter que c'est grâce à la faible sélectivité du processus de complexation par les CDs qu’il a été possible de multiplier le nombre et la diversité des molécules invitées.
Des modifications chimiques du macrocycle ont été envisagées de façon à permettre l’accroissement des constantes de complexation des complexes supramoléculaires formés par les CDs. La synthèse de cyclodextrines alkylées peut ainsi permettre le greffage "d’extensions" de cycle, comme il sera explicité dans la suite de cette étude
2.Energies mises en cause.
En solution aqueuse la cavité des CDs est occupée par des molécules d’eau énergiquement non-favorables (interaction polaire-apolaire), ce qui permet une substitution aisée par des molécules invitées de moindre polarité que l’eau.
Plusieurs contributions énergétiques ont été mises en évidence pour expliquer les interactions en jeu lors de la formation de complexes par des cyclodextrines.
C’est l’action simultanée de plusieurs de ces interactions qui rend effective l’inclusion spécifique, les phénomènes de reconnaissances moléculaires étant dus à la coopération de multiples interactions faibles. Ce phénomène d’inclusion-complexation ne fait intervenir aucune liaison covalente mais uniquement des forces telles que les liaisons hydrogène ou des interactions de Van der Waals ce qui permet ainsi le relargage de la molécule invitée, ouvrant la voie à de multiples applications.
3. Conséquences
L’inclusion d’invités dans la cavité des CDs, influence nombres de propriétés de ceux-ci, ouvrant la voie à de nombreuses études tant théoriques qu’industrielles.
Les propriétés physico-chimiques des invités se trouvent modifiées du fait de la formation de complexe d’inclusion :
La réactivité des molécules incluses est aussi modifiée lors de l’inclusion. Le plus souvent elle est diminuée car l’invité se trouve stabilisée, mais la cyclodextrine peut aussi agir comme catalyseur. La diffusion et la volatilité sont modifiées, ainsi que les propriétés chromatographiques des molécules incluses.
Le phénomène d’inclusion dans les molécules cages hydrophiles que sont les CDs, fournit, à la recherche fondamentale, un modèle d’étude des réactions enzymatiques, catalytiques, ou de complexations. De nombreuses et diverses applications des CDs sont décrites dans la littérature, ainsi que dans les registres de brevets. Toutes ces applications tirent parti des propriétés complexantes en milieu aqueux des CDs avec un panel impressionnant de molécules invitées. De nombreuses applications technologiques découlent de l’observation faite que, par la formation du complexe d’inclusion, la stabilité, la solubilité, la bio-disponibilité, la durée de vie, la toxicité et l’odeur de la molécule invitée se trouvent favorablement modifiées.
1. Complexant
La formation de ces complexes d’inclusion est mise à profit, non seulement dans le domaine pharmaceutique (solubilisation, stabilisation, masquage d’effets secondaires),mais aussi, dans les industries chimiques et agro-alimentaires53 (stabilisant d’arômes, protection des vitamines, extraction du cholestérol54). Citons aussi l’utilisation toute récente, dans des produits vaporisés sur tissus, de la CD pour y masquer les odeurs désagréables55.
2. Vectorisation.
Les cyclodextrines sont aussi employées comme vecteur56, pour le ciblage de médicaments. Quelques tentatives d'emploi des cyclodextrines dans un but de vectorisation de principes actifs ont été réalisées. Le schéma employé est identique dans toutes les approches relevées: On part de la structure de base des CDs sur laquelle on greffe chimiquement une "antenne" destinée à assurer la fonction de ciblage. L'originalité des différentes approches réside essentiellement dans la pertinence du choix de cette antenne, ainsi que dans l'approche synthétique employée lors de la réalisation de ces conjugués57.
3. Catalyseurs - modèles d'enzymes artificielles.
En plus de la sélectivité des complexes hôte-invité, les cyclodextrines possèdent des propriétés catalytiques à rapprocher de celles des enzymes (inhibitions compétitives, cinétique de Michaelis-Menten). Les CDs ont été largement étudiées en tant que modèle enzymatique de l’a-chymotrypsine, de l’anhydrase carbonique et, des ribonucléases ainsi que comme inducteur de chiralité dans des réactions aussi diverses que la réduction de ceto-acides, l’halogénation, l’oxydation de sulfures, l’époxidation, certains réarrangements sigmatropiques, l’addition de Michaël, les réactions de Diels-Alder et de Wittig, en utilisant un réactif achiral et un substrat prochiral58. Tout dernièrement, les CDs ont été utilisées comme chaperonnes artificielles pour l’étude du repliement de l’anhydrase carbonique dénaturée (utilisation séquentielle de détergents et de CDs)59.
4. Complexant chiral.
Les CDs sont aussi utilisées en séparation énantiomérique par électrophorèse capillaire, chromatographie en phase gazeuse ou chromatographie liquide haute performance60,61.
Ce sont, cependant, les applications en séparation chirale (colonnes CLHP, CPG), dans le domaine de la formulation de médicaments (galénique56) et de l’industrie agro-alimentaire qui ont donné lieu aux plus nombreuses applications des CDs. Celles-ci ont eu pour conséquences les dépôts de nombreux brevets nationaux et internationaux couvrant l’exploitation de ces applications. Les cyclodextrines ont aussi été utilisées en RMN comme auxiliaires chiraux pour la détermination d’excès énantiomériques62.
La cyclodextrine est une molécule chirale optiquement pure. En effet toutes les unités glucose sont sous une forme d. La cavité de ces CDs permet la formation de complexes d’inclusion, si le produit inclus est sous forme racémique, les complexes obtenus seront diastéréoisomères.
Il est admis qu’il faut avoir formation d’un complexe d’inclusion pour espérer observer une reconnaissance chirale par une CD. Ce qui a été vérifié en utilisant la b-CD greffée sur un support chromatographique. Aucune résolution énantiomérique n’a pu être observée avec un solvant hydrophobe, celui-ci occupant la cavité de la CD et empêchant de ce fait la formation d’un complexe d’inclusion. Il semble nécessaire que la molécule hôte doit s’adapter le plus possible à la cavité de la CD. Les molécules mal adaptées à la cavité ont un mouvement rotationnel propre ayant pour conséquence de moyenner leur position respective et donc de défavoriser leur résolution chirale. Il apparaît aussi que le centre chiral doit être proche de l’entrée de la cavité ou qu’il doit y avoir un substituant orienté de façon a avoir au moins une interaction forte avec un des hydroxyles de la CD. Quant un énantiomère répond à ces différentes conditions, il existe une forte probabilité pour avoir une reconnaissance chirale par la CD.
La problématique des conditions nécessaires à la reconnaissance chirale sera développée au chapitre suivant
2. Etudes réalisées.
Les
interactions énantiosélectives entre les molécules invitées
chirales ou prochirales et la cyclodextrine ont été étudiées
de diverses manières. Avant l’utilisation récente des CDs pour
la séparation d’énantiomères en chromatographie gazeuse,
aucun modèle théorique du mécanisme de reconnaissance chirale
n’avait été proposé. Cependant, pour la séparation
de racémiques par cristallisation fractionnée, pour les réactions
catalysées, ainsi que pour la chromatographie (en tant que phase stationnaire),
un modèle à trois-points d’interaction analogue à celui
de Easson, Stedman et Dalgliesh a été proposé. D’après
ce modèle, la partie lipophile et de préférence aromatique
de la molécule est incluse dans la cavité de la CD,
et deux substituants polaires interagissent avec les hydroxyles secondaires
de la cyclodextrine.
La cyclodextrine en tant que sélecteur chiral a fait l’objet de très nombreuses études par diverses méthodes physico-chimiques :
C. Dalgliesh74 lors d’une étude sur la résolution chirale de racémiques d’aminoacides aromatiques, sur papier chromatographique de cellulose, pose les bases de la reconnaissance chirale.
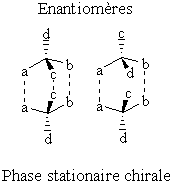
Figure 23 : Représentation schématique du modèle des trois points de Dalgliesh.
Les caractéristiques structurales citées par Dalgliesh, nécessaires pour obtenir une résolution chirale sont les suivantes :
De plus, des changements structuraux même faibles du complexant ou du complexé peuvent causer de grandes variations d’énantiosélectivité
2. Evolution du modèle des trois points.
Ce modèle n’a que très peu évolué depuis sa proposition en 1952. Les trois points d’attache sont devenus des points d’interactions. Une tendance actuelle veut que ces trois points soient un minimum nécessaire pour obtenir une séparation chirale. Toutefois, les interactions souhaitées peuvent être présentes sous forme de forces de répulsions, ce qui est un cas très fréquent. Par exemple, Shallenberger et al.75, étudièrent le modèle du récepteur "sucre" de la langue.
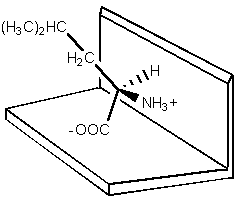 a)
a) 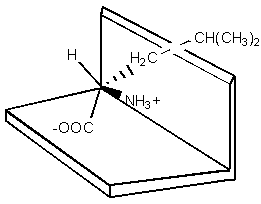 b)
b)
Figure 24 : Schéma du récepteur en présence de a) d-leucine et de b) l-leucine.
Dans le cas général, les d-amino acides peuvent se lier au récepteur en deux points, par des liaisons H, alors que cela est impossible aux dérivés l (gène stérique). Deux liaisons hydrogène et une gène stérique déterminent la stabilité de ce complexe énantiomère invité-récepteur protéinique. Il n’est pas nécessaire d’avoir trois interactions attractives entre l’hôte et l’invité pour obtenir une reconnaissance chirale, deux attractives et une répulsive sont tout aussi efficientes.
Un autre modèle de résolution chiral peut être une adaptation du modèle "clef–serrure" d’E. Fischer76.
La reconnaissance par les cyclodextrines peut être réalisée par :
1. Mécanisme "clef-serrure"
Un exemple simple de la reconnaissance chirale par des cyclodextrines, via le mécanisme "clef-serrure" est la formation de dimères chiraux du pyrène dans la cavité de la g-CD77. Le dimère formé prend préférentiellement une conformation asymétrique d’hélicité (M). Il n’y a la présence d’aucune liaison hydrogène dans le site. Un phénomène comparable est observé pour le (4)-hélicène et le 1-1’-binaphtalene, tous deux achiraux en solution. En présence de g- ou de b-CD ces composés adoptent une conformation définie (respectivement (P)-hélicité et configuration -R). Les structures fines de ces complexes ne sont pas connues mais il semble aux auteurs que la cavité de la CD joue un rôle de trou de serrure asymétrique
2. Modèle des trois points d’interactions.
Le premier exemple de reconnaissance chirale par les cyclodextrines a été décrit par Cramer et Dietsche78, il s’agit de la résolution partielle de dérivés de l’acide mandelique.
Armstrong79,80 résume ainsi les conditions nécessaires pour obtenir une séparation chirale avec des cyclodextrines, il faut :
Une molécule hétérocyclique, possédant un centre chiral dans le groupement cyclique, semble être la candidate idéale d’après les principes énoncés précédemment. L’absence d’interactions différentielles au niveau de la couronne d’hydroxyles de la cyclodextrine peut escamoter la reconnaissance chirale. L’utilisation de dérivés de cyclodextrines modifiés au niveau des hydroxyles -2 ou -3 peut fournir une solution à cette limitation.
Hamilton et Chen81,82, constatent une parfaite cohérence entre ces exigences et la résolution chirale du fenoprofen (acide 2-(3-phenoxyphenyl) propanoïque) par la b-CD, résolution étudiée par diffraction des rayons-X. Les deux isomères sont présents dans le cristal formé en présence de CD, mais chaque isomère peut être étudié de façon individuelle. L’isomère S répond à toutes ces exigences alors que le composé R n’y répond que partiellement. En effet, l’isomère S peut former d’importantes liaisons hydrogène avec les hydroxyles primaires et secondaires de la couronne tandis que l’isomère R ne le peut. Dans les cristaux, les deux isomères optent pour des arrangements différents dans le dimère de b-CD (tête-à-tête pour le R, tête-à-queue pour le S). Il en résulte une constante de complexation de la b-CD trois fois plus importante pour l’isomère S que pour l’isomère R.
Il semble que les CDs, naturelles ou modifiées, soient des hôtes présentant une faible discrimination chirale pour des invités à chiralité centrale. Mais, elles semblent être très efficaces pour des invités à chiralité axiale ou par hélicité.