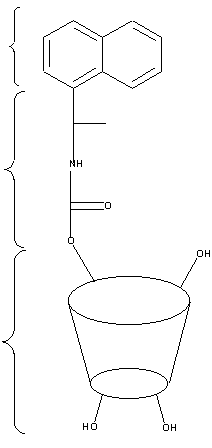
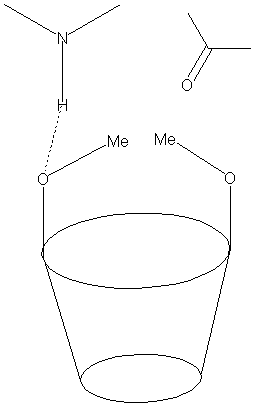
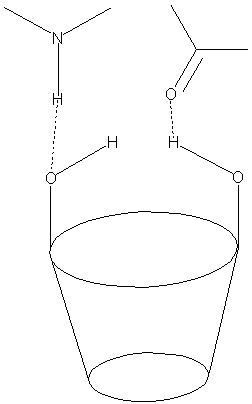
Comme explicité précédemment, il existe de façon théorique une importante corrélation entre la géométrie de complexes diastéréoisomères (formation de 3 points d’interaction) et la séparation chirale de ces différents isomères.
Cette corrélation entre la géométrie et
la thermodynamique des complexes diastéréoisomères est-elle
pratiquement observée ? Peut-on, par exemple, observer des isomères
de géométries différentes mais avec des énergies
de complexation similaires et donc ne pas obtenir de séparation physique ?
Si on observe par RMN, ou par rayons-X,
des complexes diastéréoisomères différents géométriquement
peut-on être sûr que par une méthode chromatographique reprenant
le même sélecteur chiral, il y ait séparation ? Peut-on
rechercher un sélecteur chiral pour la chromatographie par RMN ?
Les résultats sont-ils transposables ? Ces différentes interrogations
ont guidé cette recherche. Quelles techniques expérimentales utiliser
pour les évaluations thermodynamique et géométriques.
1. Méthodes expérimentales d’études thermodynamiques.
La chromatographie est une méthode qui permet de séparer les constituants d’un mélange en utilisant leurs différences de partage entre une phase mobile, dans laquelle ils sont solubles, et une phase stationnaire qui exerce sur eux un effet "attracteur". Les constituants du mélange se déplacent ainsi à des vitesses différentes sur la colonne et peuvent alors être isolés. Plusieurs types de chromatographies se distinguent :
L’étude du phénomène de complexation83
par les cyclodextrines fournit un modèle théorique d’étude
générale des phénomènes d’inclusion (tel que le
très étudié modèle
enzyme-substrat). Diverses catégories de molécules incluses ont
été étudiées en présence des cyclodextrines
naturelles, couvrant de ce fait une large gamme de classes de composés :
carbohydrates, alcools aliphatiques, amines, acides, aminoacides, dérivés
aromatiques, et de très nombreux principes actifs. Les études
portant sur des cyclodextrines modifiées recouvrent, elles, une gamme
moindre de composés.
Les principaux facteurs intervenants dans le phénomène d’inclusion font intervenir les forces de Van der Waals ainsi que des interactions hydrophobes. De même, il semble que les liaisons H et des effets stériques soient aussi des facteurs influants de la stabilité des complexes. Les données thermodynamiques obtenues expérimentalement, dans le cas de la complexation par les cyclodextrines, sont une expression des conséquences pondérées de ces divers facteurs.
Diverses méthodes sont usitées dans l’investigation thermodynamique de la complexation mais peu sont celles pouvant être utilisées dans le cadre spécifique de l’étude des séparations chirales.
Les méthodes utilisées doivent permettre une quantification
des séparations obtenues expérimentalement pour les différents
isomères. Les séparations observées sont l’expression physique
d’une différence d’énergie de stabilisation des complexes diastéréoisomères.
Il s’agit de méthodes chromatographiques.
En chromatographie liquide, les cyclodextrines sont utilisées soit dans la phase stationnaire, soit dans la phase mobile. La faible solubilité de la b-CD rend son utilisation difficile en chromatographie liquide, bien que l’ajout d’urée augmente considérablement sa solubilité ainsi que son hydrophobicité.
Les valeurs thermodynamiques sont évaluées en chromatographies gazeuse61,84 et liquide85 et, par la mesure des temps de rétention (tr) des isomères en interaction avec les cyclodextrines (greffées sur colonnes ou ajoutées dans l’éluant).
L’électrophorèse capillaire86
est utilisée dans l’étude des séparations chirales par
des CDs. Par cette méthode, il est possible, possédant des temps
de migration différents,
de déterminer les concentrations des différentes espèces
en présence (libres et complexées).
1. Chromatographie gazeuse.
Le principe de la séparation par chromatographie gazeuse (CG) consiste à partager l'échantillon à analyser entre deux phases. L'une de ces phases est un liquide stationnaire uniformément réparti sous forme d'une pellicule mince, sur un solide inerte de grande surface spécifique, tandis que l'autre phase est un gaz mobile qui s'écoule au travers de l'ensemble stationnaire.
La séparation chirale par les cyclodextrines en CG a été tout particulièrement étudiée par les équipes de Schurig3,71,87 et de König68 des études théoriques très performantes ont été réalisées par Lipkowitz et al.67.
a. Théorie.
D’après les calculs70 de Lipkowitz, la plus grande différence d’énergie d’interaction entre les deux isomères R et S existe dans la cavité du macrocycle. Il met aussi l’accent sur la symétrie interne de la cavité des cyclodextrines (d’ordre 7 pour la b-CD) qui limite la possibilité de discrimination chirale de celle-ci, bien que dans ses calculs il utilise une structure moyenne asymétrique.
b. En application pour les cyclodextrines.
Les cyclodextrines sont stables thermiquement mais les CDs naturelles sont insolubles dans nombre de solvants organiques, ce qui en limite l’utilisation pour les colonnes capillaires de CG. Les travaux précurseurs de Smolkova-Keulemansova88,89 et de Sybilska90, démontrent leur utilisation possible et efficiente en CG.
Dans nombre de publications, les cyclodextrines utilisées comme phase chirale, sont des CDs alkylées (di- et per-). L’introduction de groupements alkyl, ou alkyl-acyl, sur les différents hydroxyles des CDs permet l’obtention de dérivés à points de fusion moindres que ceux des CDs naturelles. De plus, ces dérivés restent stables thermiquement et sont solubles dans nombre de solvants organiques utilisés pour le remplissage des colonnes capillaires chromatographiques91. Les premiers travaux, dans ce domaine, ont été réalisés par l’équipe de König. L’utilisation de la b-CD perpentylée a ainsi permis la résolution de dérivés trifluoroacetylés de sucres, d’aminoacides et d’aminos alcools. La CD sembla être liquide à température ambiante mais, une étude plus poussée mit en évidence que sous une forme pure elle était cristalline. La CD utilisée pratiquement, est en fait un mélange de différents isomères ayant des degrés divers de substitution.
Les réactivités diverses des hydroxyles (2, 3, 6) des unités glucosidiques des CDs permettent de décliner les types de substituants et les degrés de substitution. Les isomères séparés par les CDs (plus ou moins dérivées) couvrent une large gamme de composés : amines, époxydes, alcools, sucres, aminoacides, stéroïdes, terpènes, ainsi que de nombreux principes actifs (anesthésiques par exemple)… Il est possible d’obtenir des énantiosélectivités inversées en changeant le groupement greffé sur la CD. Par exemple, la très polaire permethyle-O-(S)-2-hydroxypropyle-b-CD présente une sélectivité inverse de la dipentyle-b-CD vis à vis de l’arabinose92.
Il semble que les énantiosélectivités observées soient dues à l’inclusion des isomères dans la cavité des CDs, ce qui est mis en évidence par l’influence de la taille de cette cavité sur la séparation. Toutefois, des études thermodynamiques tendent, elles, à indiquer que la formation de complexes diastéréoisomères à la surface des CDs, serait responsable des séparations énantiomériques93.
Les limitations d’utilisation de la CG sont principalement dues au facteur température : l’analyte doit être volatil et à température élevée il y a des risques de racémisation.
2. Electrophorèse capillaire.
a. Théorie.
Ces dernières décennies, l’électrophorèse
capillaire a connu un fort développement comme méthode séparatrice
d’isomères, en présence de cyclodextrines comme sélecteur
chiral94. Cette technique présente l’avantage d’être
rapide, efficace, peu coûteuse en produits et d’une grande facilité
de mise en œuvre. La séparation chirale est basée sur l’addition
d’un sélecteur chiral (la CD) dans le tampon, créant ainsi un
environnement chiral dans lequel non seulement les constantes de complexation
sont différentes mais aussi les mobilités électrophorétiques
des différentes formes libres et associées des énantiomères.
Le comportement de l’analyte, lors de la migration, est décrit par trois
paramètres (la mobilité électrophorétique de l’analyte
libre, celle de complexe formé avec la CD et la constante d’équilibre
du complexe). La mobilité du complexe est dépendante des différentes
concentrations et connaît un maximum.
b. En application pour les cyclodextrines.
Les cyclodextrines utilisées peuvent être naturelles95 ou neutres électriquement, mais leur emploi est limité du fait de leur faible solubilité (en particulier de la b-CD) et de leur neutralité électrique. Pour obtenir une séparation efficiente d’isomères neutres, l’utilisation de cyclodextrines chargées semble nécessaire. Elles sont généralement modifiées (sulfate, carboxyle) de façon à obtenir un complexe chargé (donc mobile dans le champ de l’expérience). Les CDs chargées peuvent être des électrolytes faibles (limités dans leur utilisation par une gamme de pH) comme des électrolytes forts (non limités en pH). A l’instar des CDs utilisées en chromatographie gazeuse qui sont souvent sous forme de mélange mal défini de différents isomères de substitution, en électrophorèse capillaire, les CDs utilisées ne sont pas toujours parfaitement définies quant à leur substitution (degré et position des groupements). L’utilisation des CDs sous une forme de combinaison hétérogène est un sérieux handicap à la compréhension fine des mécanismes de séparation et à la prédiction de leurs résultats. Par contre, ces mélanges présentent souvent des séparations supérieures. Certaines équipes toutefois, développent des CDs modifiées parfaitement définies.
Il est important de noter que le(s) groupement(s) greffé(s) sur la cyclodextrine peu(ven)t favoriser comme défavoriser la séparation recherchée en interagissant avec l’invité.
Les limites de l’électrophorèse capillaire en
présence de CD comme sélecteur chiral, sont les limites générales
de cette technique qui n’est qu’analytique actuellement et donc,
ne peut permettre la purification des isomères étudiés.
L’étude, par cette méthode, de séparations chirales fait
apparaître la mobilité (des produits libres et complexés)
dans le champ, facteur qui n’intervient dans aucune autre technique.
Historiquement, les cyclodextrines ont tout d’abord été ajoutées à la phase mobile en chromatographie sur couche mince96 avant d’être utilisées en CLHP (Chromatographie Liquide Hautes Performances) dans l’éluant. Elles ont ensuite été incluses dans des phases stationnaires chirales du fait de leurs intéressantes propriétés de complexation, sous forme de gel de polymère97. Ces supports ne sont pas très résistants à la pression et se dégradent rapidement, c’est pourquoi de nouveaux types de supports ont été mis au point. La CD a alors été fixée à des matrices minérales par l’intermédiaire d’un bras ou d’un polymère adsorbé sur le support. De nombreuses revues retracent ces études ainsi que les résultats obtenus en CLHP80,88,98.
1. Mécanismes de séparation sur phases stationnaires chirales à base de cyclodextrines.
La séparation d’énantiomères sur phases stationnaires chirales à base de cyclodextrines repose, essentiellement, sur la formation d’un complexe d’inclusion entre la partie hydrophobe des molécules invitées que l’on souhaite séparer et la cavité apolaire de la CD. D’autres associations interviennent et permettent d’augmenter la sélectivité des supports :
Dans la plupart des cas, une combinaison de facteurs intervient, cependant, les deux premiers facteurs semblent les plus importants96 quantitativement.
L’essentiel des séparations chromatographiques
s’effectue en mode polarité de phases inverse (mélanges méthanol/eau
ou acétonitrile/eau, souvent additionnés d’un tampon).
En mode polarité de phases inverse, les composés les plus retenus sont les moins polaires.
Les molécules de CD sont très utilisées en CLHP sous forme naturelle ou dérivée, en solution dans la phase mobile, greffées sur de la silice (ou sur des matrices organiques) ou encore incluses dans des polymères adsorbés sur des matrices minérales. Grâce à leur propriété de complexation en polarité de phases inversée, les CDs ont permis la séparation de nombreux composés aromatiques : isomères géométriques ou de positions, énantiomères, composés pharmaceutiques…
D’autres séparations s’effectuent en
polarité de phases normale (phase mobile à base de mélange
d’hydrocarbure [souvent de l’hexane] et d’alcool [souvent de l’isopropanol]).
Dans ce dernier cas l’énantiosélectivité s’appuie sur la
formation de liaisons H, de liaisons de Van der Waals et d’associations dipolaires
avec les groupements hydroxyles placés à l’extérieur de
celle-ci99 et, non plus sur la complexation
de la molécule avec la cavité. En chromatographie à polarité
de phases normale, les composés les plus retenus sont les plus polaires.
Un troisième type de phase mobile est apparu récemment, appelé phase organique polaire. Il s’agit d’un mélange d’acétonitrile, de méthanol, d’acide acétique et de triéthylamine. Comme précédemment l’énantiosélectivité semble être obtenue par l’intermédiaire d’associations entre le soluté et l’extérieur des molécules de b-CD, tandis que l’intérieur est occupé par l’éluant.
Les CDs sont utilisées en mode normal et en mode organique polaire lorsqu’elles sont dérivées (acétylées, benzoylées, carbamatées, hydroxypropylées…). Dans ces cas là, le mécanisme de séparation est basé sur des interactions se produisant à l’extérieur de la cage de la cyclodextrine, il n’y a plus de complexation. Ces supports ont permis la séparation de nouveaux composés, parfois non séparés sur les supports natifs. Ils mettent en jeu des mécanismes de séparation complexes encore mal connus.
Néanmoins, le mécanisme principal de séparation d’un grand nombre d’isomères repose sur la formation d’un complexe d’inclusion. La séparation des composés exploite la différence de constante de complexation des complexes formés avec la cavité hydrophobe. Le composé le plus retenu correspond au complexe d’inclusion le plus stable.
a. Chromatographie sur phases stationnaires chirales à base de dérivés de cyclodextrines.
Les groupements hydroxyles secondaires de la b-CD naturelle sont relativement fixes dans l’espace. Leurs modifications chimiques permettent l’introduction d’une certaine flexibilité aux bords de la cage. De nouveaux supports ont ainsi été préparés.
Les groupes hydroxyles en position -2 peuvent être fonctionnalisés avec un certain nombre de substituants, susceptibles de modifier l’hydrophobicité et la sélectivité des phases. L’ensemble de leurs propriétés physico-chimiques et de leurs capacités de complexation peut aussi bien se trouver bouleversé.
Beaucoup de séparations chirales ont été
effectuées en polarité de phases normale sur des phases greffées
de b-CD naturelle100
mais, le mécanisme de séparation d’énantiomères
en mode normal n’a pas été décrit. A l’inverse, 4 phases
greffées de b-CD substituées (acétylée,
N-diméthylephénylecarbamate, N-1-napht-1-yléthylcarbamate,
p-toluyester) ont été utilisées avec succès
dans ces conditions101,102,103. Par substitution,
à l’aide de groupements fonctionnels aromatiques ou carbonylés,
les possibilités d’association p-p
sont accrues,
avec de concert l’existence d’associations avec les groupements hydroxyles résiduels
de la b-CD. De ce fait, chaque phase stationnaire
dérivée présente une énantiosélectivité
spécifique104.
Certains racémates séparés sur phases greffées de dérivés de b-CD en mode normal, sont également résolus sur les phases à bases de CD naturelles en polarité de phases inverse. Toutefois, les séparations en polarité de phases normale sont réalisées avec des temps de rétention plus réduits et une meilleure résolution qu’en polarité de phases inverse101. De manière générale, les dérivés de la CD utilisés ne sont pas complètement substitués. Nombre d’études portent d’ailleurs, sur l’influence du degré de substitution de la CD vis-à-vis de la rétention observée. Ces différentes études semblent indiquer un lien entre la rétention et le degré de substitution.
b. Cyclodextrines acétylées en chromatographie liquide.
Les premiers essais de modification chimique aux extrémités des cavités de CD ont conduit à l’acétylation des hydroxyles des CDs (par analogie avec les celluloses triacétylées). Les phases obtenues sont très hydrophobes, les possibilités de formation de liaisons H sont affectées (par suppression du pouvoir donneur de "proton").
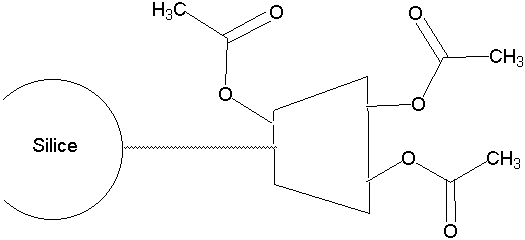
Figure 25 : Support à base de cyclodextrines acétylées.
Des modifications de la profondeur de la cavité de la
CD, de sa flexibilité, de son accessibilité et de sa capacité
à former des liaisons H avec les solutés peuvent être supposées.
De nouvelles associations sont susceptibles d’apparaître avec les groupements
acétylés et de modifier sensiblement l’énantiosélectivité
des colonnes. Mais, celle-ci semble inférieure à celle des phases
stationnaires de CDs naturelles. Nonobstant, certains composés tels que
le Norgestrel ne peuvent être séparés que sur cette famille
de support104. Un certain nombre d’aminoacides
dansylés ont été séparés sur les supports
acétylés de Tanaka et al. alors que sur Cyclobond I AC (Cyclobond
acétylée) aucun de ces composés n’était séparé105.
Certains composés phénoliques ainsi que des alcools et des amines
sont susceptibles d être plus retenus et mieux résolus que sur
les supports natifs. Les phases acétylées sont recommandées
dans le cas d’alcools ou d’amines aromatiques, sur lesquelles le centre asymétrique
se trouve en position a ou b.
c. Phases stationnaires chirales à base d’ester paratoluyle de b-cyclodextrine.
Ce type de support à base de CDs substituées par des groupements toluyle ester possède des sites accepteurs de liaisons H (liés à la présence du carbonyle) ainsi que des groupements hydroxyles résiduels susceptibles de servir de sites de complexation. Ces supports semblent adaptés pour des composés comportant des groupements carbonylés ou aminés tels que la N-CBZ, (dl) proline ou le Phensuximide101.
d. Phases stationnaires chirales à base de cyclodextrines benzoylées.
Ces supports à base de CDs benzoylées106, obtenus par Tanaka et al., permettent la séparation des isomères de position d’un certain nombre de composés aromatiques bifonctionnels (crésol, toluidine…) ainsi que celle d’un mélange de composés anti-épileptiques (primidone, phénobarbitol, carbamazépine et phénytoïne).
e. Phases stationnaires chirales à base de cyclodextrines propionylées.
Tanaka et al. ont également synthétisé des supports à base de cyclodextrines propionylées106 qui ont permis la séparation d’isomères de position de benzènes substitués. Néanmoins, les séparations sont moins efficaces que celles obtenues sur les phases à base de CDs benzoylées.
f. Phases stationnaires chirales à base de b-cyclodextrines modifiées par des groupements carbamates.
Le fait qu’un groupement isocyanate soit fixé sur une
cage de CD, elle-même fixée à de la silice crée un
environnement chiral complexe qui permet de nombreuses séparations.
Le support est obtenu par réaction de la CD, fixée à la
silice avec un isocyanate101.
|
Phase mobile |
Interaction |
Site d’interaction |
|
|
Phase
normale
|
Interaction p-p |
Atome
chiral du Naphtyléthyle carbamate (NEC)
|
|
|
Phase
organique polaire
|
Complexe
chiral
|
Site
donneur/accepteur de liaisons H
|
|
|
Phase
inverse
|
Complexe
d’inclusion
|
Molécules
chirales de glucose
|
Figure 26 : Support à base de (R) ou de (S)-Naphtylcarbamate de CD.
Ce type de support a été surnommé "phase stationnaire chirale multimodale" car il est susceptible d’être utilisé efficacement avec trois types de phases mobiles, grâce à l’intervention de trois modes d’actions distincts. Ainsi la résolution de composés inséparables sur des phases naturelles a été obtenue. Le choix de l’éluant est fait en fonction de la structure, de la solubilité et de la stabilité du composé étudié.
Les meilleurs résultats des colonnes sont obtenus en
mode polarité de phases normale. Ces phases s’apparentent alors aux supports
de type Pirkle. Le mode d’action des autres dérivés carbamatés
de la b-CD est analogue à celui du 1-napht-1-yléthylcarbamate,
mais ce dernier semble le plus stéréosélectif103.
De plus, la forme (R) du N-naphtyléthyle carbamate confère au
support une sélectivité différente de sa forme (S) et de
sa forme racémique102. Par exemple, les dérivés
d’aminoacides sont presque toujours mieux résolus sur les colonnes
(R)-NEC, tandis qu’environ 75% des composés racémiques testés
sont mieux résolus sur les colonnes (S)-NEC.
Des énantiomères d’aminoacides dansylés ont été résolus en polarité de phases inverse sur supports à base de phénylcarbamate de b-CD et de propylcarbamate de b-CD107. Ces deux supports, bien que de structures proches, ont montré une sélectivité différente. Ce qui confirme l’importance des associations à surface des cages dans le mode de séparation.
g.Phases stationnaires chirales à base de cyclodextrines (S) ou (R,S) hydroxypropylées.
Les groupements hydroxyles de la cyclodextrine réagissent avec le (S) ou le (R,S) oxyde de propylène afin d’obtenir des dérivés (S) ou (R,S) hydroxypropyles éther fixés à une silice glycidoxy sililée.
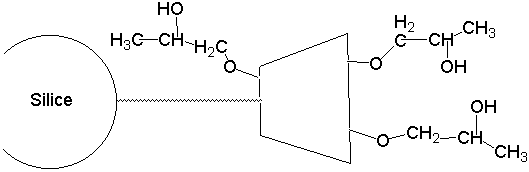
Figure 27 : Supports à base de CDs polyhydroxypropylées.
La substitution des hydroxyles par des groupements hydroxypropyles, a comme conséquence de permettre une plus grande distance entre le centre chiral et le site de liaisons H potentielles. La phase de CDs substituées par des groupements (R,S) hydroxypropyle apparaît comme une phase stationnaire d’application générale. Elle permet la séparation d’un certain nombre de composés non aromatiques tels que les dérivés N-tertiobutoxycarbonyle aminoacides108. Le composé (S) montre une sélectivité accrue dans certaines séparations, telle la méthadone.
Le mode d’action de ce type de phase repose sur l’inclusion des composés dans la cage et sur la formation d’associations entre le composé étudié et les groupements hydroxypropyles. De plus, la présence d’un centre chiral supplémentaire sur le groupement greffé peut contribuer à l’énantiosélectivité par la formation de liaisons H stéréospécifiques. Ce phénomène est particulièrement important dans le cas de molécules trop volumineuses pour s’inclure profondément dans la cavité.
h. Phases à base de cyclodextrines méthylées.
La société Macherey Nagel a commercialisé un support à base de CDs perméthylées fixées à de la silice Nucléosil sous le nom de Nucléodex b PM. Sur ce support, tous les groupements hydroxyles ont été remplacés par des groupements méthoxyles, modifiant ainsi les caractéristiques de sélectivité des colonnes.
|
|
|
|
Accepteur de proton seulement Nucleodex bPM |
Donneur et accepteur de protons Nucleodex bOH |
Figure 28 : Comparaison des dispositions possibles des liaisons H sur supports natif et méthylé.
Un fait particulièrement remarquable concerne les temps de rétention observés sur ce type de phases qui se trouvent diminués pour un même éluant. La perméthylation de la CD modifie le nombre et le type d’associations possibles : l’hydrophobie de l’extrémité de la cage est accrue. De plus, la possibilité de formation des liaisons H est considérablement abaissée.
La phase méthylée possède uniquement des propriétés basiques dues aux paires d’électrons libres de l’oxygène. Cette propriété permet une meilleure séparation, plus rapide, de certains composés tels que le méphorbarbital. Pour ce composé, les associations avec les hydroxyles de la CD naturelle conduisent à des associations supplémentaires qui allongent les temps de rétention sans pour autant améliorer la sélectivité. Cette phase stationnaire méthylée permet de séparer des composés non résolus sur les phases à base de CD naturelles tels que les dérivés du méthylmécoprop. Néanmoins, des composés, qui nécessitent des groupements hydroxyles libres pour être résolus (la chlortalidone par exemple), ne sont pas séparés sur les phases totalement méthylées. En polarité de phases inverse, un certain nombre de stéroïdes, d’acides carboxyliques, d’amides et d’imides carboxyliques ont été résolus sur les colonnes Nucleodex b PM. Néanmoins, une perte d’efficacité des colonnes a été relevée vis-à-vis des dérivés aromatiques disubstitués, tandis que certains principes actifs et dérivés du naphtalène ont été résolus sur des colonnes à bases de cyclodextrines méthylées109.
i. Conclusion.
Les phases stationnaires chirales dérivées de CDs présentent plusieurs avantages notables : leur stabilité en phase aqueuse rend possible leur utilisation pour l’analyse de fluides biologiques, leur coût est moindre comparé à celui des phases stationnaires du type I. Elles conviennent bien à l’analyse de routine, et leur énantiosélectivité ainsi que leur sensibilité élevées permettent l’analyse de traces. En contrepartie, leur efficacité est en moyenne inférieure de 20% à celles des phases stationnaires classiques, ce qui nuit à la qualité de la résolution. Aucune de ces phases stationnaires n’est universelle. Chacune d’entre elles présente des spécificités et des déficiences.
Il peut être intéressant de connaître finement la géométrie des complexes (diastéréoisomères ou non) formés avec les cyclodextrines. En effet, une connaissance fine des géométries peut permettre une meilleure compréhension des différents paramètres intervenant dans les mécanismes de complexation, un des objectifs pouvant être l’amélioration des constantes de complexation par fonctionnalisation spécifique de la CD.
Certains complexes de CDs ont pu être obtenus sous
une forme mono-cristalline et analysés par diffraction des rayons-X,
permettant ainsi la connaissance de leur structure.
Les complexes diastéréoisomères peuvent être formés
séparément ou en en présence du racémique dans la
solution de cristallisation.
Les CDs naturelles et fonctionnalisées ont fait l’objet de différentes études, principalement par Harata et al.65,66,110, en présence d’invités chiraux ou non. Cette équipe a tout particulièrement étudié des cyclodextrines per-méthylées en présence de différents isomères optiques.
Cette méthode d’étude géométrique donne des résultats très fins permettant de distinguer les liaisons H mises en jeu dans la stabilisation des complexes. Lorsque les deux isomères ont pu être cristallisés en présence de CD, il est alors aisé d’observer les différences structurelles des complexes diastéréoisomères81 formés. Il est aussi possible d’identifier certaines interactions stabilisantes, expliquant partiellement certaines différenciations observées par d’autres méthodes.
Toutefois, cette méthode est fortement limitée par la possibilité (ou non) d’obtention de mono-cristaux de complexes, de taille suffisante pour l’analyse. De plus, les complexes étudiés sont par définition en phase solide et peuvent différés géométriquement de ceux effectivement présents en solution, d’où une difficulté pour corréler les résultats, avec ceux obtenus en solutions (chromatographie par exemple).
Dans le cas des complexes d’inclusion, la résonance magnétique nucléaire (RMN) présente par rapport à d’autres techniques, la possibilité d’observer simultanément en solution, les deux espèces, complexant et complexé, en interaction. Elle permet de situer avec précision le(s) lieu(x) de l’interaction et, par l’observation simultanée des deux molécules, de mettre en évidence, toutes modifications chimiques ou conformationnelles de l’une ou l’autre des deux espèces.
La formation de complexes diastéréoisomères entraîne la formation d’environnements magnétiques différents pour chaque isomère comme pour le complexant par rapport aux mêmes molécules libres. Il est possible de supposer que les environnements chimiques étant différents, les spectres RMN de chacun des complexes diastéréoisomères le seront. Lors de l’inclusion dans la CD, du fait de la chiralité de la cavité, les deux énantiomères forment des complexes diastéréoisomères. L’environnement magnétique de chacun des énantiomères se trouve alors modifié différemment par la CD. Cette différence d’environnement magnétique, se traduit en RMN par des différences de déplacements chimiques. Il est possible alors de visualiser, si les différences d’environnement sont importantes, un dédoublement des signaux du mélange des deux énantiomères, ainsi qu’une variation des déplacements chimiques des protons de la CD et plus particulièrement des protons de la cavité.
2.Informations sur la complexation par inclusion dans la cyclodextrine.
Les avantages de cette technique spectroscopique par rapport aux autres méthodes sont nombreux : ainsi elle permet de mettre en évidence la spécificité de la formation d'un complexe (observation d’interactions entre la molécule organique invitée et la cavité), les caractéristiques du complexe (stœchiométrie et constante de complexation du complexe formé), d'avoir plusieurs mesures indépendantes (grâce à la multiplicité des signaux 1H-RMN d'une même molécule organique), d'éviter les erreurs d’interprétation dues à la présence d'impuretés, etc...
Les principales techniques de RMN mises en œuvre reposent sur l'observation des noyaux 1H (grâce à son fort rapport gyromagnétique et de son abondance naturelle), bien que des études reposant sur l’observation des noyaux 13C soient aussi rapportées.
Le principe de base de l’étude par RMN des complexes d'inclusions CDs-invités repose sur l'étude des variations des valeurs de déplacements chimiques (d en ppm) des signaux de la molécule invitée et/ou de la molécule hôte. De ce fait, en présence d'un complexe, les modifications de la densité électronique locale à l’intérieur de la cavité sont créées par la molécule invitée, entraînant des variations des déplacements chimiques de certains protons des CDs. Les protons H3 et H5 situés à l'intérieur de la cavité sont les principales victimes de ces modifications d’environnement électronique.
Ce phénomène est particulièrement accentué lorsque les molécules invitées présentent des insaturations, sources d'anisotropie locale de densité électronique.
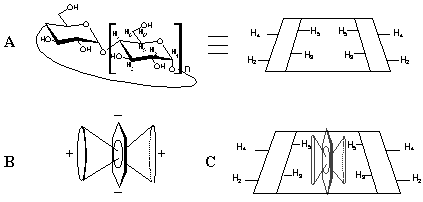
Figure 29: A: Représentation schématique d'une CD et localisation des divers protons du macrocycle; B: Représentation du cône d'anisotropie créé par la délocalisation des électrons d'un cycle aromatique; C: Influence de ce cône sur les protons H3 et H5 situés à l’intérieur de la cavité.
Cependant, l'observation des variations de déplacements chimiques ne permet d'obtenir qu'un nombre limité d'informations concernant la partie de la molécule mise en jeu dans le processus d'inclusion.
3. Etudes des interactions dipolaires dans des complexes : les séquences NOESY
et ROESY.
La séquence 2D NOESY (Nuclear Overhauser Effect SpectroscopY), permet d'étudier la relaxation spin-réseau longitudinale simultanément pour tous les protons présents dans une molécule.
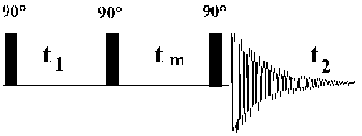
Figure 30: La séquence 2D NOESY.
Il est possible, en utilisant cette séquence, d’avoir
accès aux termes de relaxation croisée homonucléaire de
chacun des noyaux et, de mettre en évidence des proximités spatiales
inter nucléaires (< 5 Å). Ces noyaux peuvent être portés
par la même molécule,
ou bien, par deux molécules différentes, comme entre une molécule
invitée et une molécule hôte. Cette séquence est
composée de trois impulsions de 90°:
On relève dans la littérature quelques exemples d'études de complexes d'inclusion de CDs mis en évidence par la séquence NOESY. Mais, la majorité des complexes formés avec les CDs en solution aqueuse, à une température proche de l'ambiante et, étudiés à haut champ (w0 ~ 500 MHz), présente une valeur de temps de corrélation tc telle que l'effet nOe est proche de la nullité (w0tc ~ 1.1).
Cet inconvénient de la séquence NOESY peut néanmoins être contourné en utilisant la séquence ROESY (Rotating frame Overhauser Effect SpectroscopY). La séquence ROESY permet d'étudier la relaxation croisée non plus selon Oz comme dans le cas du NOESY, mais selon l'axe Oy. Ceci est réalisé après avoir "bloqué" les aimantations, selon Oy à l'aide d'un champ radiofréquence B1 continu et suffisamment intense que l’on applique au centre du spectre (champ de "spin-lock") afin de conserver les aimantations alignées selon cet axe.
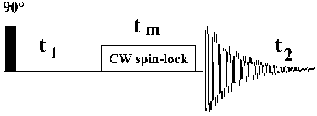
Figure 31: La séquence 2D ROESY de base.
L'expression des couplages dipolaires a alors lieu comme dans la séquence NOESY durant le temps de mélange tm, mais sous l'influence du champ radiofréquence B1 et selon l'axe Oy du référentiel tournant. L'observation de ces couplages est réalisée juste à la fin de la période de mélange, après la coupure de B1.
Il est remarqué ainsi que quelle que soit la valeur
de w0tc
, s'is reste toujours positif:
On observe donc toujours des signaux hors-diagonaux résultant des couplages
dipolaires.
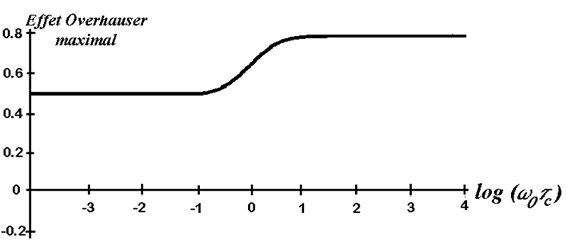
Figure 32: Effet Overhauser nucléaire maximal dans le cas du ROESY en fonction de log (w0tc).
L'emploi de cette séquence en mode phasé permet aussi de différencier les pics de corrélation rOe des pics dus aux échanges chimiques, de signes opposés. Un autre avantage de cette séquence réside dans le fait que les phénomènes de diffusions de spins y sont fortement atténués.
Ainsi, l'utilisation de cette séquence pour l’étude des complexes de CDs naturelles ou modifiées permet de lever les ambiguïtés quant aux noyaux mis en jeu dans le processus d'inclusion. L’observation de pics de corrélations rOe entre les protons de la molécule hôte et ceux de la molécule invitée permettra de montrer les proximités spatiales intermoléculaires, et donc, de mettre en évidence la formation du complexe.
D'une façon générale, l’observation de pics de corrélations dipolaires dans un spectre ROESY 2D d'un mélange, permet de montrer sans ambiguïté la formation d’un complexe d’inclusion en solution. Cependant, les données structurales obtenues à l'aide de cette séquence ne suffisent pas à caractériser totalement le complexe formé. Les paramètres thermodynamiques qui régissent l’équilibre du complexe en solution doivent encore être déterminés et, la mise en œuvre d'expériences de RMN 1D permet de déterminer ces inconnues. Un effet secondaire du ROESY étant le transfert du relais, lorsque des informations fines seront nécessaires des expériences T-ROESY seront utilisées111.
4. Conclusion.
L’étude par RMN 1D permet une visualisation rapide du phénomène d’inclusion dans les cyclodextrines quand celui-ci a lieu. Lorsque les CDs sont mises en présence d’un racémique, s’il y a formation de complexes d’inclusions, les complexes formés sont alors diastéréoisomères. Les environnements chimiques des différents protons (de la CD et de l’invité) sont donc modifiés et les spectres obtenus lors de la complexation sont différents de ceux des produits isolés. De plus, du fait de la diastéréoisomérie des complexes formés, les spectres peuvent être différents. Les complexes possèdent des géométries internes différentes et donc des environnements magnétiques différents pour certains protons. Point qui se traduit par un dédoublement de certains pics des protons concernés. Par RMN, il est donc possible de visualiser rapidement une différence de géométrie des complexes. Une étude des proximités spatiales par RMN bidimensionnelle peut permettre de connaître finement les géométries de complexes en positionnant l’invité dans la cavité de la CD.
Toutefois, les dédoublements sont généralement visibles uniquement sur la molécule invitée. Le spectre de la cyclodextrine représente donc une moyenne entre les formes complexées avec le racémique et la forme libre. Ce qui peut présenter, par la suite, une difficulté pour connaître finement les structures des différents complexes en présence.
De plus, des différences d’environnement magnétique peuvent être faibles et donc se traduire par des spectres faiblement dédoublés (voir non dédoublés, suivant la résolution de l’expérience RMN), bien que les complexes puissent avoir des géométries très différentes globalement.
Une autre limite, est que les signaux des protons de la CD couvrent une grande région de spectre (région élargie avec des cyclodextrines non symétriques), et peut donc dissimuler de nombreux protons de la molécule invitée.
La présence d’un phénomène d’inclusion
est en général démontrée sans ambiguïté
par la comparaison du spectre proton de la cyclodextrine seule avec celui du
complexe.
Les différences de déplacements chimiques entre les deux sont
alors liées à l'inclusion d'une molécule à l'intérieur
de la cavité de la CD. En effet, la molécule invitée entraîne
des modifications de l'environnement de son hôte, lesquelles se traduisent
par des variations de déplacements chimiques (d)
dues essentiellement aux effets d'anisotropie, induits par des groupes carbonyles
ou des cycles aromatiques par exemple. Sur la CD ce sont les déplacements
chimiques des protons H3 et H5
situés à l'intérieur de la cavité qui sont le plus
fortement modifiés. Cette observation de la modification des spectres
RMN ne permet en revanche aucune évaluation directe de la constante d’inclusion.
Le calcul de la constante de complexation peut être réalisé à partir de différents spectres RMN 1D, par la méthode dite de Job. Les constantes obtenues représentent une approche des paramètres thermodynamiques de la séparation.
La complexation d'une (ou de plusieurs) molécule(s) invitée(s) par une cyclodextrine est décrite par un équilibre qui suit la loi d'action de masse, et qui est le suivant :
Où [A], [B] sont respectivement les concentrations de A et B sous forme libre et [C] la concentration du complexe pur.
|
A |
+ B |
|
(1) |
|
|
concentration à t0 |
A0 |
B0 |
A = cyclodextrine |
|
|
concentration à l’équilibre |
A0-c |
B0-c |
B = molécule invitée |
|
|
A(B)n = Complexe 1:n formé |
||||
|
c = Avancement de la réaction en M-1 |
A l'équilibre thermodynamique, c = [A(B)n]eq, et donc:
[A]eq=(At-[A(B)n]eq) et [B]eq=(Bt-n[A(B)n]eq).
Ka peut donc être réécrit sous la forme:
|
|
(2) |
On définit par k (en Hz) la constante
de vitesse globale de l'équilibre et qui équivaut
à k1+k-1.
On note dl
le déplacement chimique d'un des protons de la molécule
invitée à l'état libre (le raisonnement reste identique
si un proton de l’hôte est pris en considération),
dc le déplacement
chimique de ce même proton à l'état complexé,
et on pose Dd = dl
- dc
(en Hz).
Lors de l’étude par RMN du proton du phénomène de complexation à l’équilibre thermodynamique, on se trouve confronté à deux cas:
Si Dd >> k, le système est dit en échange lent à l'échelle de temps d’observation de la RMN, pour une fréquence donnée (500 MHz dans notre cas) : deux types de signaux séparés seront observés pour chacun des deux états des molécules: les signaux des produits libres et les signaux des produits complexés (dc et di).
Si Dd << k, le système est dit en échange rapide à l'échelle de temps d’observation de la RMN, pour une fréquence donnée (500 MHz dans notre cas) : un seul type de signaux pour chacune des molécules présentes sera observé, résultant de la moyenne des signaux des produits à l’état libre et des produits à l’état complexé (dobs).
Ce phénomène est représenté schématiquement ainsi:
Figure 33: Illustration de l'influence de la vitesse d’échange sur la forme des raies et sur les valeurs de déplacements chimiques des signaux obtenus en RMN.
La majorité des complexes d'inclusion obtenus avec les cyclodextrines donne lieu, lorsque l’étude de ces derniers est effectuée à haut champ, à température ambiante et en milieu aqueux, à un échange rapide, en RMN du proton. Il est observé, en effet, une variation des valeurs de déplacements chimiques des signaux des protons situés à l'intérieur de la cavité (H3 et H5) sous l’influence de la molécule invitée, qui résulte d’un signal moyen entre les déplacements chimiques de la forme libre et de la forme complexée des protons H3 et H5.
Cette situation d’échange rapide ne permet pas d'avoir accès directement à la stœchiométrie du complexe. Pour ce faire, il faut mettre en œuvre des expériences de titration, appelées aussi méthode de Job.
Cette méthode, encore appelée méthode
des variations continues permet de déterminer le coefficient
stœchiométrique d'un complexe en équilibre, en absence
de tout autre phénomène compétitif d’association,
en observant l’évolution d’une variable
physico-chimique au cours du dosage. La RMN-1H est utilisée
comme méthode physico-chimique d’investigation en suivant les
restrictions imposées par cette méthode, la variable observée
étant le déplacement chimique moyen dobs
(ppm) des protons.
L'accès à n (coefficient stœchiométrique du complexe formé, voir (1)) requiert de préparer une gamme de mélanges des deux composés A et B à volume constant, toutes les entités devant être totalement hydrosolubles, en conservant constante la quantité A0 + B0, et en faisant varier le ratio r défini par :
|
r = A0 / (A0+B0) = A0 / C (r = fraction molaire initiale de A) |
(3) |
En posant :
|
C = A0+B0 = cste |
(4) |
Dans ce cas, en utilisant les équations (3) et (4), les concentrations des diverses espèces à l'équilibre s'écrivent :
|
[A]eq = A0 - [A(B)n]eq = rC - [A(B)n]eq |
(5) |
|
[B]eq = B0 - n[A(B)n]eq = C(1-r) - n[A(B)n]eq |
(6) |
Pour une valeur de ratio r donné, la concentration en complexe à l'équilibre [A(B)n]eq atteint un maximum pour lequel la dérivée d[A(B)n]eq/dr s'annule. Ainsi, les dérivées partielles des équations (1), (5) et (6) par rapport à r, à ce maximum, conduisent aux égalités suivantes :
|
d(1)/dr Þ [B]eq.d([A]eq)/dr + n[A]eq.d([B]eq)/dr = dK/dr = 0 |
(7) |
(car K est indépendant de At et Bt, à température fixée).
|
d(5)/dr Þ d([A]eq)/dr = C |
(8) |
|
d(6)/dr Þ d([B]eq)/dr = -C |
(9) |
En remplaçant les égalités (8) et (9) dans (7), on aboutit donc à l'égalité suivante :
|
[B]eq.C - n[A]eqC = 0 soit [B]eq = n[A]eq |
(10) |
En remplaçant cette valeur dans (6), on obtient alors :
|
n[A]eq = C(1-r) - n[A(B)n]eq |
(11) |
Egalité qui peut être injectée dans (5), afin d’obtenir :
|
(C(1-r) - n[A(B)n]eq )/n = rC - [A(B)n]eq |
(12) |
Donc, au maximum, en réarrangeant l’égalité (12), on obtient la condition suivante :
|
à l'équilibre thermodynamique r = 1/(1+n) " K, " C |
(13) |
Lorsque la RMN du proton est utilisée comme
méthode d’observation, dans le cas d'un échange rapide,
une valeur de déplacement chimique dobs
est observée pour n'importe quel proton donné, de B ou
de A, qui est une valeur moyenne du déplacement chimique dl
du composé libre (à l'état pur), et
dc du composé
complexé (à l'état pur). Ce déplacement
chimique dobs est pondéré
par la concentration à l'équilibre de chacune des deux
espèces
(libre et complexée). Ceci se traduit par l'égalité,
pour un proton H donné de A (ou d’un proton de B), du
type :
|
dobs (H)At = dc (H)[A(B)n]eq + dl (H)[A]eq |
(14) |
Ddobs et Ddc sont introduits, étant respectivement l'écart de déplacement chimique pour un proton donné entre le signal observé (dobs(H)) et le signal de l'espèce libre (dl(H)), et l'écart de déplacement chimique entre l'état libre et complexé (dc(H)), ce qui se traduit, pour une proportion de A et B donnée :
|
Ddobs(H) = dobs(H) - dl(H) |
(15) |
|
Ddc(H) = dc(H) - dl(H) |
(16) |
La substitution de (15) et (16) dans (14) permet d'obtenir l'équation (17), pour un proton H donné de la CD :
|
Ddobs (H)At = Ddc (H)[A(B)n]eq |
(17) |
Une relation entre AtDdobs (H) et [A(B)n]eq est obtenue, c'est à dire, entre Ddobs (H) et n. Ainsi, le tracé du produit AtDdobs(H), en fonction de r pour un proton donné (lorsque le système est à l'équilibre thermodynamique, en présence d’un seul type de complexe 1:n et dans le cas d'un phénomène de complexation pur sans autre phénomène d’association compétitif) présente un maximum à la valeur :
r = 1 / (1+n) (13)
Il reste cependant encore à déterminer la constante d'association Ka du complexe formé, sous les mêmes conditions expérimentales, afin de compléter la caractérisation thermodynamique du complexe formé.
Dans le cas d'un échange lent, la concentration de complexe ([A(B)n]eq) formé est facilement accessible (lorsque n = 1) par l'intégration des signaux des protons du complexe : At et Bt étant connus, il est alors facile d'accéder à la valeur de K en remplaçant ces valeurs déterminées par intégration dans l'expression (2) de K.
Dans le cas d'un échange rapide, cette constante n’est pas directement accessible. Une des approches possibles est d'utiliser la méthode développée par Benesi et Hildebrand. Cette méthode permet de déterminer graphiquement K en utilisant les données
de la RMN
(ou bien, tout autre méthode physico-chimique), dans le cas d'un complexe 1:1, sous la restriction que le composé non observé soit en large excès
(» facteur 10) par rapport à l'espèce observée.
Une autre approche, moins restrictive quant aux conditions expérimentales, consiste à utiliser les données acquises pour obtenir les tracés de Job, et de les traiter mathématiquement.
L’expression générale est :
Où A, B, C représentent respectivement un isomère, la cyclodextrine et le complexe A(B)n.
Dans cette expression n représente le coefficient stœchiométrique de K la constante d’association.
Cette constante est définie ainsi
|
(18) |
La relation (14) restant toujours valable, pour un proton donné et observé de la molécule hôte (HC) ou pour un proton donné de la molécule invitée (HB).
Dans le cas d’un complexe de stœchiométrie 1 :1 (n=1), il peut être déterminé la valeur de la constante d’association en utilisant la méthode de Bénési-Hildebrand modifiée112.
En RMN deux cas peuvent se présenter :
|
(19) |
Où dAobs, dAl et dAc représentent respectivement la valeur du paramètre observé et ses valeurs à l’état libre et dans le complexe pur.
La combinaison des équations (1), (2), (3) et (4) donne après quelques calculs algébriques une seule équation pour l’espèce A :
|
(20) |
Cette équation est donc constituée
de trois variables observables (Ddobs, At et Bt),
et de deux inconnues (K et Ddc). Les expériences réalisées précédemment, afin de calculer la stœchiométrie du complexe, permettent d’accéder à une série
de valeurs de ces variables observables (Ddobs, At et Bt).
Ces données expérimentales peuvent donc être traitées mathématiquement à l’aide d'une procédure de régression des moindres carrés multiparamétriques afin d’accéder aux valeurs calculées
des deux inconnues, K et Ddc.
Un programme réalisé au laboratoire permet d'effectuer cette opération dans le cas d'une stœchiométrie 1:1. Le programme peut être utilisé sans aucune restriction concernant les concentrations des deux espèces et, il converge rapidement avec précision
vers les valeurs de K et de Ddc, Ddc correspondant à la valeur maximale de la variation de déplacement chimique du protons (entre la forme libre et la forme complexée
pure).
Cette procédure nécessite la valeur de paramètre à l’état libre, c’est-à-dire les valeurs de déplacement chimique de la molécule invitée dans l’eau.
La constante d’association
apparente K pouvant être déterminée à partir de n’importe quel signal de la molécule incluse, l’utilisation de toutes les données obtenues pour tous les déplacements
chimiques donne en fin de compte une valeur moyenne de la constante K.
Ce calcul peut être mené sur la cyclodextrine et sur la molécule
incluse.
Cet algorithme
de calcul peut être appliqué à chacun des protons des deux molécules formant le complexe, l’homogénéité des résultats obtenus pour chacun des différents protons étant, de la même façon, un critère de pertinence du résultat. Il est à noter que ce traitement mathématique n'est valable qu’en présence
d’un seul type de complexe (dans ce cas, 1:1),
sans aucune autre interaction compétitive (micellisation, agrégation...).
En effet, cette procédure de calcul n'est applicable que dans le cas d'un phénomène de complexation unique. Or, la présence de toute autre interaction intermoléculaire perturbe elle aussi les valeurs de déplacements chimiques, qui ne sont alors plus uniquement fonction du phénomène de complexation avec la CD. La présence de ce phénomène d’interaction compétitif biaise donc les données brutes, empêchant la convergence de la régression.
Pour tenter d’évaluer la différence de constante de complexation entre deux énantiomères vis-à-vis de la cavité de la d (différence provenant de structure entre les deux complexes formés), le calcul des constantes d’associations semble être une méthode rapide et efficace. Toutefois, une difficulté nouvelle se présente, les équations décrites auparavant, ne concernent que la formation non compétitive d’un complexe. Dans le cadre de cette étude, il y a formation compétitive de deux complexes diastéréoisomères : deux molécules incluses se trouvent en solution et ce de fait il y a les deux isomères. La situation est donc plus complexe et la procédure précédente ne peut convenir pour calculer les deux constantes d’association qui se trouvent être en compétition. Une modification du calcul a opérer doit être réalisée pour refléter le phénomène compétitif.
Cette difficulté de prise en compte de complexations compétitives, peut être facilement levée en étudiant la formation des complexes séparément pour chaque isomère. Dans le cas des racémiques, la disponibilité d’énantiomères optiquement purs n’est pas toujours. Cependant il est possible d’ecrire de nouvelles équations dans le cas de la complexation compétitive pour chaque énantiomère.
Où B représente la cyclodextrine, Enx représente un des isomères et Cpxx le complexe BEnx. Kx la constante d’association se définit ainsi :
|
(21) |
Où [B], [Enx] sont respectivement les concentrations de B et Enx sous forme libre et [Cpxx] la concentration du complexe.
La complexation est compétitive vis-à-vis de B, on a donc :
|
(22) |
1 et 2 sont relatif à chaque isomère
Il est possible d’en déduire alors que :
|
(23) |
|
|
(28) |
Dans le cas de l’échange rapide, pour chaque isomère, on observe une valeur moyenne de d:
|
(24) |
Où dEnxobs, dEnxlibre et dEnxc représentent respectivement les valeurs du déplacement chimique observé et ses valeurs à l’état libre et dans le complexe pur pour Enx.
Pour simplifier la notation il est posé :
Et
D’où l’on peut déduire que :
|
(25) |
Pour simplifier l’écriture des équations, on pose :
Les équations deviennent alors :
|
(26) |
|
|
(27) |
Pour simplifier les calculs, on se place dans les conditions d’un mélange racémique en En1 et En2 d’où :
L’équation finale obtenue est la suivante :
|
(28) |
||
|
(29) |
||
Cette équation est à rapprocher de celle obtenue dans le cas non compétitif
|
(24) |
Une procédure similaire de régression à multiples paramètres peut donc être utilisée.
Ddobs pour En1 et En2, [B]t et [En]t, étant connus, il faut effectuer le calcul une évaluation de Y (ou de Z). Un calcul itératif de Y et de Z va être effectué. Pour ce faire, les mesures de l’évolution des Ddobs vont être utilisées et il est procédé à une extrapolation de la valeur attendue de dc pour chaque isomère, ce qui donne accès aux valeurs approchées de Z (et de Y ) correspondantes.
Ces valeurs sont soumises, ainsi que celles déterminées par les mesures sur le spectre (Ddobs, [B]t et [En]t), au programme de régression. Le programme calcule K et Ddc de l’énantiomère concerné. Le calcul est alors effectué pour l’autre énantiomère en utilisant de Ddc calculé précédemment comme base de calcul pour Y (ou Z). On effectue ainsi plusieurs cycles de calcul pour obtenir une valeur constante de Ddc pour chacun des isomères.
Cette procédure n’ayant pas encore, à notre connaissance été utilisée, il faut la valider. Ainsi, des mesures de K ont été réalisées pour deux complexes dans des conditions compétitives et ont été comparées à celles obtenues lors de complexations non compétitives.
Un premier test de validation a été réalisé en reprenant les données d’une inclusion non compétitive et en les utilisant dans le nouveau programme, la valeur du Dd2obs/Dd2c étant alors mise à zéro (le deuxième isomère n’existant pas). Le programme donne les mêmes valeurs que celles du calcul classique. Ce test n’est toutefois pas suffisant pour valider ce programme, une étude dans les conditions de la compétition est indispensable.
Il faut donc choisir deux molécules s’incluant dans une même cyclodextrine (dont les K respectives sont connues) et n’interagissant pas entre elles. De nombreux produits s’incluant dans des CDs ont été étudiés, mais il en existe peu répondant à toutes ces conditions. Pour contourner la difficulté, le problème a été inversé, la validation de la méthode a été effectuée sur la complexation compétitive de deux CDs : un produit s’incluant dans deux CDs différentes à été retenu pour cela. L’option s’est portée sur l’indométacine dont les constantes d’inclusion dans la b- et la g-CD sont connues113.
Figure 34 : Structure de l’indométacine.
Une série de solutions de concentrations variables en indométacine en b- et en g-CD a été réalisée, tout en conservant la concentration de la b-CD égale à celle de la g-CD (dans les calculs, l’hypothèse d’un mélange équimolaire en En1 et En2 ayant été posée).

Figure 35 : Spectre (1H, D2O, 500MHz, 298K) d’une solution 5 mM en b-CD et en g-CD et
10mM en indométacine.
Sur ce spectre, on peut observer que les H1 des deux CDs sont séparés, mais tous les autres signaux des protons sont entremêlés ; il est donc impossible d’y relever les différentes variations de déplacements chimiques pour les deux CDs. Pour effectuer les mesures indispensables au calcul de constantes, le recours à une expérience RMN multidimensionnelle TOCSY a été nécessaire, permettant les transferts de l’aimantation des H1 vers les différents protons de la CD. Ainsi, il est possible de visualiser sans difficulté une grande partie des déplacements chimiques de chacune des CDs.

Figure 36 : Spectre TOCSY (1H, D2O, 500 MHz, 298K, temps demélange 80ms) d’une solution à 6 mM en b-CD et en g-CD et, 14mM en indométacine.
Les variations de déplacements chimiques des différents protons de chaque cyclodextrine ont été relevées et utilisées pour déterminer la valeur de chacune des constantes d’association à l’aide du programme décrit précédemment.

Figure 37 : Variations des d des H1 de la b-CD et de la g-CD en présence de concentrations variables d’indométacine. a) 8/8/4 mM, b) 6/6/6 mM c) 5/5/10 mM d) 4/4/12mM e) 2/2/16mM
Les valeurs moyennes obtenues sont du même ordre de grandeur que celles déterminées pour chaque complexe seul.
Tableau 3 : Valeurs des constantes obtenues en présence de complexation compétitive et sans.
|
Complexation compétitive |
Complexation non compétitive |
La méthode de calcul précédemment exposée se trouve ainsi validée. Elle permet de calculer la différence de constante de complexation de chaque énantiomère vis-à-vis de la cavité de cyclodextrines naturelles ou modifiées. De plus, elle donne accès aux paramètres RMN de chaque complexe pur. Cette méthode peut être généralisée au calcul de n’importe quel paramètre variant avec l’inclusion (fluorescence…). Au demeurant, elle peut être utilisée pour des mélanges non équimolaires en compétiteur, il faut de ce fait pondérer la valeur Y (et Z) du facteur de rapport des concentrations. Ce qui peut permettre, lorsque les produits à étudier sont en petites quantités, d’évaluer simultanément les constantes d’inclusion dans deux CDs. Il faut, toutefois, être uniquement en présence des deux complexations de stœchiométrie 1:1
Le principe de la méthode de Job, de calcul de constantes de complexation, repose sur une variation des déplacements chimiques en fonction des concentrations relatives du complexant et du complexé. Pour calculer les constantes de complexation respectives d’un racémique en présence de cyclodextrine, il faut pouvoir relever indépendamment chaque déplacement chimique.
Le déplacement chimique observé est fonction du déplacement du complexe pur, de la constante de complexation, et du rapport des concentrations complexant/complexé.
Pour ce qui est de la séparation chirale, la valeur importante en CLHP est le rapport des constantes de complexation. En effet, c’est ce rapport qui régit les différences de temps de rétention entre les isomères, et donc leur éventuelle séparation.
Plusieurs cas peuvent théoriquement se présenter :
Les calculs des variations des déplacements chimiques des protons dédoublés sont réalisés suivant les cas évoqués. Une représentation graphique est utilisée pour visualiser aisément les différentes données calculées.
Le premier cas envisagé est le cas où les complexes ont des constantes de complexation proches et des dc proches aussi. Il est aisé de se rendre compte que dans ce cas, la visualisation de la discrimination chirale par l’observation de pics dédoublés sera peu probable comme représenté ci-dessous. Les constantes de complexations choisies pour le calcul sont de 1000 M-1 et 1100 M-1, les variations des déplacements chimiques des complexes purs sont de 10 et 10.5 (unité arbitraire que l’on peut considérer d’une valeur de 0.1ppm), avec un dlibre de 0. Les calculs des déplacements chimiques des complexes en fonction des concentrations des différentes espèces sont réalisés et représentés graphiquement pour la clarté du propos.

Figure
38 : Variations relatives des déplacements chimiques observés
en fonction du pourcentage de complexe formé, pour deux complexes
de K et de dc très
proches
(les largeurs de pics utilisées sont arbitraires).
Ce cas peut correspondre à la complexation non stéréospécifique de diastéréoisomères, lorsque le site inclus de la CD est très éloigné du C* par exemple.
L’hypothèse choisie ici, est le cas de complexes diastéréoisomères ayant des valeurs de déplacements chimiques très différentes pour des constantes de complexation identiques. La variation des déplacements chimiques est représentée ci-dessous. Les constantes de complexation sont fixées à 1000 M-1 pour les deux espèces, les déplacements chimiques des complexes libres sont de 10 et 15 avec dlibre de 0. Les calculs des déplacements chimiques des complexes en fonction des concentrations des différentes espèces sont réalisés et représentés graphiquement.

Figure 39 : Variations relatives des déplacements chimiques observés en fonction du pourcentage de complexe formé, pour deux complexes de même K et de dc très différents
(les largeurs de pics utilisées sont arbitraires).
Par la représentation, ci-dessus, des résultats du calcul des variations des déplacements chimiques pour chacun des complexes formés, il est aisé de déduire qu’une discrimination chirale peut être observée par RMN même si les constantes de complexation sont similaires. Ce qui importe ce sont les valeurs différentes de dc pour chacun des complexes purs. Une très grande différence de valeur de dc entre chacun des complexes permet une échelle de variation du déplacement chimique observée très importante et de ce fait, un dédoublement des pics pour les complexes diastéréoisomères formés. Dans ce cas, théorique, il y a discrimination chirale (observation de dédoublement de pic) mais pas de séparation (les K étant considérées dans ce calcul comme identiques).
Le dernier cas théorique à explorer tant au niveau de la discrimination qu’au niveau de la séparation chirale est celui où le rapport entre les constantes est important alors que la différence des déplacements chimiques des complexes purs est faible (facteur géométrique). Les déplacements chimiques des complexes purs sont de 10 et 11 (dlibre=0)avec des constantes de complexation de 1000 et 5000 M-1 reciproquement.

Figure 40 : Variations relatives des déplacements chimiques observés en fonction du pourcentage de complexe formé, pour deux complexes de dc proches et de K très différentes
(les largeurs de pics utilisées sont arbitraires).
Il est visible sur cette représentation graphique que par RMN, la discrimination chirale ne pourra être observée que pour des taux importants de complexation.
Ces calculs théoriques mettent en évidence une différence entre discrimination chirale et séparation chirale. De façon théorique, l’observation d’une différenciation chirale n’entraîne pas automatiquement l’observation de l’autre différenciation chirale pour les mêmes conditions expérimentales.
L’étude des paramètres thermodynamiques de la différenciation chirale se fait par des expériences chromatographiques. La chromatographie liquide permet l’utilisation de nombreuses cyclodextrines comme sélecteur chiral. Les CDs utilisées lors des études relevées dans la littérature, sont souvent chimiquement modifiées. Les différentes modifications chimiques relevées ne sont généralement pas complètement contrôlées, ce qui fait apparaître une grande hétérogénéité quant aux cyclodextrines résultantes. Il s’agit, le plus souvent, d’un mélange de CDs modifiées à des degrés divers sur des positions pas systématiquement contrôlées. Cette diversité des CDs présentes a pour conséquence une difficulté de reproductibilité des résultats entre les différents lots de CD utilisés lors du geffage sur colonne.
L’étude des paramètres géométriques de la différenciation chirale se fait par des expériences de diffraction des rayons-X ou par RMN. Cette dernière méthode présentant les avantages de s’intéresser aux espèces en solution et une grande facilité de mise en œuvre. Dans le cas de l’étude par RMN, les cyclodextrines utilisées sont parfaitement décrites et contrôlées lorsqu’elles sont modifiées. La RMN peut permettre un accès aux constantes de complexation sous certaines conditions.
Lors de l’étude bibliographique, malgré la richesse sur le sujet, il n’est pas apparu d’analyses comparatives sur des cyclodextrines communes entre les méthodes chromatographiques et géométriques. Une recherche reprenant les mêmes cyclodextrines pour les analyses par RMN et par CLHP peut permettre une compréhension plus fine des différents paramètres, thermodynamiques et géométriques, intervenant dans la différenciation chirale.